| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Principles of Therapeutics
Overview
|
The regulations governing the development of new drugs have evolved over the past century to assure the safety and efficacy of new medications for the population. The safety or efficacy of a drug in an individual patient is never assured. Because all patients differ in their responses to drugs, each therapeutic encounter must be considered an experiment with a hypothesis that can be tested. The scientific basis of the hypothesis derives from the database generated from controlled clinical trials during drug development and the experience obtained postmarketing. Well-defined endpoints must be established prior to therapy. These may be clinical endpoints, such as reduction of fever or pain, or they may be surrogate markers, such as reduction of blood cholesterol or blood pressure, that are correlated with the clinical outcome. Individualization of therapy for a particular patient requires a basic understanding of pharmacokinetics and pharmacodynamics. Many factors can influence that patient's response to a drug, including the age of the patient; disease of the organs of drug elimination (kidney, liver); the concurrent use of other drugs, foods, and chemicals (drug interactions); previous therapy with the same or similar drugs (tolerance); and a variety of genetic factors that can influence the kinetics and toxicity of drugs (pharmacogenetics). For a limited number of drugs, monitoring of the concentration of the drug in plasma can be useful to control for pharmacokinetic variability. Monitoring of pharmacodynamic variability requires close attention to the patient's responses, using predefined goals for acceptable efficacy and toxicity. Some adverse events are extensions of the drug's pharmacological effect and often are avoidable if therapy is individualized. However, other serious adverse reactions are related to an interaction of the drug with variables unique to the individual patient. When a drug is first marketed, it has been tested in only a limited number of well-characterized patients. Adverse events that occur as commonly as 1 per 1000 patients may not be discovered prior to marketing, and rare events may not be discovered for several years after a drug is on the market. It is the responsibility of all health care professionals to monitor the effects of drugs postmarketing and to report serious adverse events that may be drug-related to the FDA and/or the drug manufacturer. In the future, it is likely that the genetic and environmental bases of interindividual variation and of rare, adverse drug reactions will be discovered and that screening techniques will be applied to individualize therapy and assess individual risk. This would improve the overall safety of pharmacotherapy. |
Therapy as a Science
|
Over a century ago, Claude Bernard formalized criteria for gathering valid information in experimental medicine. However, application of these criteria to therapeutics and to the process of making decisions about therapeutics has, until recently, been slow and inconsistent. Although the diagnostic aspects of medicine are approached with sophistication, therapeutic decisions often are made on the basis of impressions and traditions. Over the past three decades, the principles of human experimentation have been defined, and the techniques for evaluation of therapeutic interventions have progressed to the point that it should now be considered absolutely unethical to apply the art, as opposed to the science, of therapeutics to any patient who directly (the adult or child) or indirectly (the fetus) receives drugs for therapeutic purposes. Therapeutics must now be dominated by objective evaluation of an adequate base of factual knowledge. This philosophy has been popularized recently under the terminology of 'evidence-based medicine.' Conceptual Barriers to Therapeutics as a Science The most important barrier that inhibited the development of therapeutics as a science seems to have been the belief that multiple variables in diseases and in the effects of drugs are uncontrollable. If this were true, the scientific method would not be applicable to the study of pharmacotherapy. In fact, therapeutics is the aspect of patient care that is most amenable to the acquisition of useful data, since it involves an intervention and provides an opportunity to observe a response. Recently, it has become evident that many of the important aspects of disease cannot be adequately assessed by objective data. For instance, dyspnea may not be predicted by measures of pulmonary function with spirometry, and the pain of angina pectoris often is not well correlated with ST-segment depression on the electrocardiogram. Nonetheless, there is a tendency to ignore or denigrate the subjective, symptomatic 'soft' data in favor of the objective 'hard' endpoints. The challenge for clinical investigators has been to objectify or 'harden' subjective measures so that they are useful for quantifying drug responses. It is now appreciated that clinical phenomena that are important to patientssuch as dyspnea, pain, and ability to functioncan be defined, described, and quantified with some precision. The approach to complex clinical data has been artfully discussed by Feinstein (1983, 1999). Another barrier to the realization of therapeutics as a science was overreliance on traditional diagnostic labels for disease. This encouraged the physician to think of a disease as static rather than dynamic, to view patients with the same 'label' as a homogeneous rather than a heterogeneous population, and to consider a disease as a single entity even when information about pathogenesis was not available. If diseases are not considered to be dynamic, 'standard' therapies in 'standard' doses will be the order of the day; decisions will be reflexive. Needed instead is an attitude that makes the physician responsible for recognition of and compensation for changes that occur in pathophysiology as the underlying process evolves. For example, the term myocardial infarction refers to localized destruction of myocardial cells caused by interruption of the blood supply; however, decisions about therapy must take into account a variety of autonomic, hemodynamic, and electrophysiological variables that change as a function of the time, size, and location of the infarction. Failure to take all such variables into account while planning a therapeutic maneuver may result in ineffective therapy in some patients while exposing others to avoidable toxicity. A diagnosis or label of a disease or syndrome usually indicates a spectrum of possible causes and outcomes. Therapeutic experiments that fail to control for the known variables that affect prognosis yield uninterpretable data. Often, if not usually, not all of the relevant variables are known. In such cases, the response to a therapeutic intervention can be a clue to sort out the parameters that contribute to the response and may be a way to discover the underlying variables that contribute to the disease. A third conceptual barrier was the incorrect notion that data derived empirically are useless, because they are not generated by application of the scientific method. Empiricism often is defined as the practice of medicine founded on mere experience, without the aid of science or a knowledge of principles. The connotations of this definition are misleading; empirical observations need not be scientifically unsound. In fact, concepts of therapeutics have been greatly advanced by the clinical observer who makes careful and controlled observations of the outcome of a therapeutic intervention. The results, even when the mechanisms of disease and their interactions with the effects of drugs are not understood, are nevertheless often crucial to appropriate therapeutic decisions. Frequently, the initial suggestion that a drug may be efficacious in one condition arises from careful, empirical observations that are made while the drug is being used for another purpose. Examples of valid empirical observations that have resulted in new uses of drugs include the use of penicillamine to treat arthritis, lidocaine to treat cardiac arrhythmias, propranolol and clonidine to treat hypertension, and sildenafil for male erectile dysfunction. Conversely, empiricism, when not coupled with appropriate observational methods and statistical techniques, often results in findings that are invalid or misleading. Clinical Trials Application of the scientific method to experimental therapeutics is exemplified by a well-designed and well-executed clinical trial. Clinical trials form the basis for therapeutic decisions by all physicians, and it is therefore essential that they be able to evaluate the results and conclusions of such trials critically. To maximize the likelihood that useful information will result from the experiment, testable hypotheses of the study must be clearly defined, homogeneous populations of patients must be selected, appropriate control groups must be found, meaningful and sensitive indices of drug effects must be chosen for observation, and the observations must be converted into data and then into valid conclusions. The sine qua non of any clinical trial is its controls. Many different types of controls may be used, and the term controlled clinical trial is not synonymous with randomized, double-blind, placebo-controlled trial. Selection of a proper control group is as critical to the eventual utility of an experiment as the selection of the experimental group. Although the randomized, double-blind controlled trial is the most effective design for avoiding bias and distributing unknown variables between the 'treatment' and 'control' groups, it is not necessarily the optimal design for all studies. It may be impossible to use this design to study disorders that occur rarely, disorders in patients who cannotby regulation, ethics, or bothbe studied (e.g., children, fetuses, or some patients with psychiatric diseases), or disorders with a typically fatal outcome (e.g., rabies), where historical controls can be used. There are several requirements in the design of clinical trials to
test the relative effects of alternative therapies. (1) Specific outcomes
of therapy that are clinically relevant and quantifiable must be measured.
These may include subjective assessments, which are important in determining
whether a therapy improves the patient's well-being. Quality of life can be
assessed by the experimental subject and can be tabulated objectively and
incorporated into evaluation of a therapy (Guyatt et al., 1993).
Wherever possible, well-defined clinical endpoints, i.e., survival or
pain relief, should be used, rather than an intermediate endpoint or
'surrogate' marker (Fleming and DeMets, 1996; Bucher et al.,
1999). A surrogate marker is a clinical sign or laboratory test that
correlates with the clinical outcome of a disease. Blood pressure, blood
cholesterol, CD4 lymphocyte count in acquired immunodeficiency syndrome (AIDS),
and premature ventricular complexes are examples of surrogate markers that
have been used as endpoints in clinical trials. Although surrogate markers
often are useful to reduce the length and sample size of a clinical trial,
the results of such trials may be misleading, as the Cardiac Arrhythmia
Suppression Trial (CAST) demonstrated (Echt et al., 1991). In CAST,
the antiarrhythmic drugs encainide, flecainide, and moricizine
were effective in suppressing ventricular arrhythmias (the surrogate marker)
in patients following a myocardial infarction, but the drugs nonetheless
increased mortality. The ultimate test of a drug's efficacy must rest with
actual clinical outcomes. (2) The accuracy of diagnosis and the severity
of the disease must be comparable in the groups being contrasted;
otherwise, false-positive and false-negative errors may occur. This is a
particular issue in developing therapies for poorly understood syndromes,
such as fibromyalgia and chronic fatigue syndrome. (3) The dosages of
the drugs must be chosen and individualized in a manner that allows relative
efficacy to be compared at equivalent toxicities or allows relative
toxicities to be compared at equivalent efficacies. (4) Placebo effects,
which occur in a large percentage of patients, can confound many
studiesparticularly those that involve subjective responses; controls must
take this into account ( The results of clinical trials of new therapeutic agents or of old agents for new indications may have severe limitations in terms of what can be expected of drugs when they are used in an office practice (Feinstein, 1994). To reduce variability, patients for experimental trials often are selected to eliminate coexisting diseases and concomitant therapy. Such trials usually assess the effect of only one or two drugs, not the many that might be given to or taken by the same patient under the care of a physician. Clinical trials usually are performed with relatively small numbers of patients for periods of time that may be shorter than are necessary in practice, and compliance may be better controlled than it can be in practice. These factors lead to several inescapable conclusions:
|
Individualization of Drug Therapy
|
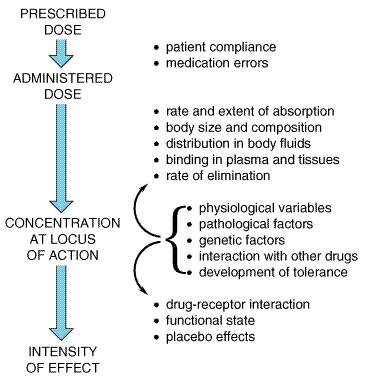
As has been implied above, therapy as a science does not apply simply to the evaluation and testing of new, investigational drugs in animals and human beings. It applies with equal importance to the treatment of each patient as an individual. Therapists of every type have long recognized and acknowledged that individual patients show wide variability in response to the same drug or treatment method. Progress has been made in identifying the sources of variability. Important factors are presented in Figure 31; the basic principles that underlie these sources of variability have been presented in Chapters 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination and 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect. The following discussion relates to the strategies that have been developed to deal with variability in the clinical setting. (See also Appendix II.)
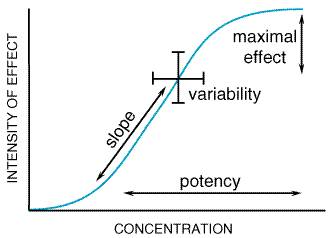
Pharmacokinetic Considerations Interpatient and intrapatient variation in disposition of a drug must be taken into account in choosing a drug regimen. For a given drug, there may be wide variation in its pharmacokinetic properties among individuals. For some drugs, this variability may account for one-half or more of the total variation in eventual response. The relative importance of the many factors that contribute to these differences depends in part on the drug itself and on its usual route of elimination. Drugs that are excreted primarily unchanged by the kidney tend to have smaller differences in disposition among patients with similar renal function than do drugs that are inactivated by metabolism. Of drugs that are extensively metabolized, those with high metabolic clearance and large presystemic (first-pass) elimination have marked differences in bioavailability, whereas those with slower biotransformation tend to have the largest variation in elimination rates among individuals. Studies in identical and nonidentical twins have revealed that genotype is a very important determinant of differences in the rates of metabolism (Penno and Vesell, 1983). For many drugs, physiological and pathological variations in organ function are major determinants of their rate of disposition. For example, the clearance of digoxin and gentamicin is related to the rate of glomerular filtration, whereas that of lidocaine and propranolol is dependent primarily on the rate of hepatic blood flow. The effect of diseases that involve the kidneys or liver is to impair elimination and to increase the variability in the disposition of drugs. In such settings, measurements of concentrations of drugs in biological fluids can be used to assist in the individualization of drug therapy. Since old age and renal or hepatic diseases also may affect the responsiveness of target tissues (e.g., the brain), the physician should be alert to the possibility of a shift in the range of therapeutic concentrations. A test should not be performed simply because an assay is available. More assays of drugs are available than are generally useful. Determinations of concentrations of drug in blood, serum, or plasma are particularly useful when well-defined criteria are fulfilled: (1) There must be a demonstrated relationship between the concentration of the drug in plasma and the eventual therapeutic effect that is desired and/or the toxic effect that must be avoided. (2) There should be substantial interpatient variability in disposition of the drug (and small intrapatient variation). Otherwise, concentrations of drug in plasma could be predicted adequately from dose alone. (3) It should be difficult to monitor intended or unintended effects of the drug. Whenever clinical effects or minor toxicity are measured easily (e.g., the effect of a drug on blood pressure or blood coagulation), such assessments should be preferred in the decision to make any necessary adjustment of dosage of the drug. However, the effects of some drugs in certain settings are not easily monitored. For example, the effect of Li+ on manic-depressive illness may be delayed and difficult to quantify. For some drugs, the initial manifestation of toxicity may be serious (e.g., digitalis-induced arrhythmias or theophylline-induced seizures). The same concepts apply to a number of agents used for cancer chemotherapy. Other drugs (e.g., antiarrhythmic agents) produce toxic effects that mimic symptoms or signs of the disease being treated. Many drugs are used for prophylaxis of an intermittent, potentially dangerous event; examples include anticonvulsants and antiarrhythmic agents. In each of these situations, titration of drug dosage may be aided by measurements of concentrations of the drug in blood (4). The concentration of drug required to produce therapeutic effects should be close to the value that causes substantial toxicity (see below). If this circumstance does not apply, patients could simply be given the largest dose known to be necessary to treat a disorder, as is commonly done with penicillin. However, if there is an overlap in the concentrationresponse relationship for desirable and undesirable effects of the drug, as is true for theophylline, determinations of concentration of drug in plasma may allow the dose to be optimized. All four of the above-described criteria should be met if the measurement of drug concentrations is to be of significant value in the adjustment of dosage. Knowledge of concentrations of drugs in plasma or urine also is particularly useful for the detection of therapeutic failures that are due to lack of patient compliance with a medical regimen or for identification of patients with unexpected extremes in the rate of drug disposition. Assay of drugs to assist the physician in achieving a desired concentration of drug in blood or plasma (i.e., 'targeting' the dose) is another example of the use of an intermediate or surrogate endpoint of therapy in place of the ultimate clinical goal. Surrogate markers also can be applied in other ways; one is to provide an indication for a change in the choice of drug therapy. Measurements of concentrations of drugs in plasma and/or measurements of one or more pharmacological effects of the drug can provide an indication of probable lack of efficacy. Other issues of importance with regard to the measurement and interpretation of drug concentrations are discussed in Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination and Appendix II. Pharmacodynamic Considerations Considerable interindividual variation in the response to drugs remains after the concentration of the drug in plasma has been adjusted to a target value; for some drugs, this pharmacodynamic variability accounts for much of the total variation in responsiveness among patients. As discussed in Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect, the relationship between the concentration of a drug and the magnitude of the observed response may be complex, even when responses are measured in simplified systems in vitro, although typical sigmoidal concentrationeffect curves usually are seen (see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect). When drugs are administered to patients, however, there is no single characteristic relationship between the drug concentration in plasma and the measured effect; the concentrationeffect curve may be concave upward, concave downward, linear, sigmoid, or an shape. Moreover, the concentrationeffect relationship may be distorted if the response being measured is a composite of several effects, such as the change in blood pressure produced by a combination of cardiac, vascular, and reflex effects. However, such a composite concentrationeffect curve often can be resolved into simpler curves for each of its components. These simplified concentrationeffect relationships, regardless of their exact shape, can be viewed as having four characteristic variables: potency, slope, maximal efficacy, and individual variation. These are illustrated in Figure 32 for the common sigmoidal log doseeffect curve.
Potency The location of the concentrationeffect curve along the concentration axis is an expression of the potency of a drug. Although often related to the dose of a drug required to produce an effect, potency is more properly related to the concentration of the drug in plasma to approximate more closely the situation in isolated systems in vitro and to avoid the complicating factors of pharmacokinetic variables. Although potency obviously affects drug dosage, potency per se is relatively unimportant in the clinical use of drugs as long as the required dose can be given conveniently and there is no toxicity related to the chemical structure of the drug rather than to its mechanism. There is no justification for the view that more potent drugs are superior therapeutic agents. However, if the drug is to be administered by transdermal absorption, a highly potent drug is required, since the capacity of the skin to absorb drugs is limited. Maximal Efficacy The maximal effect that can be produced by a drug is its maximal, or clinical, efficacy (which is related to, but not precisely the same as, the term efficacy as discussed in Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect). Maximal efficacy is determined principally by the properties of the drug and its receptoreffector system and is reflected in the plateau of the concentrationeffect curve. In clinical use, however, a drug's dosage may be limited by undesired effects, and the true maximal efficacy of the drug may not be achievable. The maximal efficacy of a drug is clearly a major characteristicof much greater clinical importance than its potency. Furthermore, the two properties are not related and should not be confused. For instance, although some thiazide diuretics have similar or greater potency than the loop diuretic furosemide, the maximal efficacy of furosemide is considerably greater. Slope The slope of the concentrationeffect curve reflects the mechanism of action of a drug, including the shape of the curve that describes drug binding to its receptor (see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect). The steepness of the curve dictates the range of doses that are useful for achieving a clinical effect. Aside from this fact, the slope of the concentrationeffect curve has more theoretical than practical usefulness. Biological Variability Different individuals vary in the magnitude of their response to the same concentration of a single drug or to similar drugs when the appropriate correction has been made for differences in potency, maximal efficacy, and slope. In fact, a single individual may not always respond in the same way to the same concentration of drug. A concentrationeffect curve applies only to a single individual at one time or to an average individual. The intersecting brackets in Figure 32 indicate that an effect of varying intensity will occur in different individuals at a specified concentration of a drug or that a range of concentrations is required to produce an effect of specified intensity in all of the patients. Attempts have been made to define and measure individual
'sensitivity' to drugs in the clinical setting, and progress has
been made in understanding some of the determinants of sensitivity to drugs
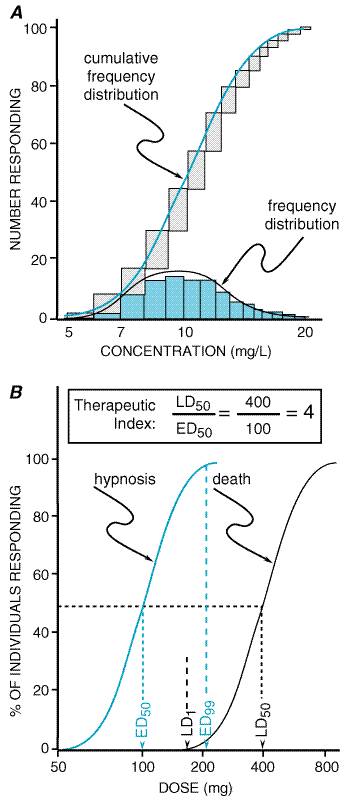
that act at specific receptors. For example, responsiveness to ConcentrationPercent or Quantal ConcentrationEffect Curve The concentration of a drug that produces a specified effect in a single patient is termed the individual effective concentration. This is a quantal response, since the defined effect is either present or absent. Individual effective concentrations usually are lognormally distributed, which means that a normal variation curve is the result of plotting the logarithms of the concentration against the frequency of patients achieving the defined effect (Figure 33A). A cumulative frequency distribution of individuals achieving the defined effect as a function of drug concentration is the concentrationpercent curve or the quantal concentrationeffect curve. This curve resembles the sigmoid shape of the graded concentrationeffect curve discussed above (Figure 32), but the slope of the concentrationpercent curve is an expression of the pharmacodynamic variability in the population rather than an expression of the concentration range from a threshold to a maximal effect in the individual patient.
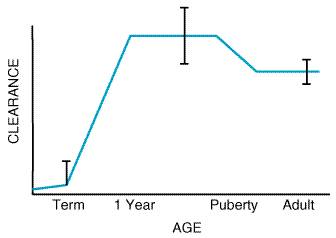
The dose of a drug required to produce a specified effect in 50% of the population is the median effective dose, abbreviated as the ED50 (Figure 33B). In preclinical studies of drugs, the median lethal dose, as determined in experimental animals, is abbreviated as LD50. The ratio of the LD50 to the ED50 is an indication of the therapeutic index, which is a statement of how selective the drug is in producing its desired versus its adverse effects. In clinical studies, the dose, or preferably the concentration, of a drug required to produce toxic effects can be compared to the concentration required for the therapeutic effects in the population to evaluate the clinical therapeutic index. However, since pharmacodynamic variation in the population may be marked, the concentration or dose of drug required to produce a therapeutic effect in most of the population will usually overlap the concentration required to produce toxicity in some of the population, even though the drug's therapeutic index in an individual patient may be large. Also, the concentrationpercent curves for efficacy and toxicity need not be parallel, adding yet another complexity to the determination of the therapeutic index in patients. Finally, no drug produces a single effect, and, depending on the effect being measured, the therapeutic index for a drug will vary. For example, much less codeine is required for cough suppression than for control of pain in 50% of the population, and thus the margin of safety, selectivity, or therapeutic index of codeine is much greater as an antitussive than as an analgesic. Other Factors That Affect Therapeutic Outcome The variation in pharmacokinetic and pharmacodynamic parameters that accounts for much of the need to individualize therapy has been discussed. Other factors, listed in Figure 31, also should be considered as potential determinants of success or failure of therapy. The following presentation serves as an introduction to these subjects, some of which also are discussed in Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination and Appendix II. Age Most drugs are developed and tested in young to middle-aged adults. At each extreme of the age spectrum, individuals differ both in the way they handle drugs (pharmacokinetics) and in their response to drugs (pharmacodynamics). These differences may require substantial alterations in the dose or dose regimen to produce the desired effect in the young or in the very old. Children Most medications have not been developed or specifically evaluated in children, and formulations often are inadequate for proper administration. Thus, development of new drugs for children and rational use of old compounds require an integrated approach to pharmacokinetic, pharmacodynamic, and formulation issues. There is no reliable, broadly applicable principle or formula for converting doses of drugs used in adults to doses that are safe and effective in children. When the drug manufacturer does not provide adequate information about pediatric dosage, there can be substantial risk in deriving a dose for children and infants from an adult dose by, for example, simply reducing the dose based upon body weight or surface area. In general, pathways of drug clearance (hepatic and renal) are limited in the newborn, particularly the premature infant. The unique physiology of the newborn has led to past therapeutic disasters, such as gray-baby syndrome (inadequate glucuronidation of chloramphenicol with drug accumulation) and sulfonamide-induced kernicterus (displacement of bilirubin from plasma proteins in the face of increased bilirubin production from fetal erythrocyte turnover, decreased bilirubin conjugation, acidosis, and decreased blood-brain barrier). Careful pharmacokinetic studies in the newborn coupled with clinical therapeutic drug monitoring have markedly improved our knowledge of neonatal developmental pharmacology and resulted in safe therapeutics. Pathways of drug clearance develop variably over the first year of life and may be influenced by induction of drug-metabolizing enzymes (e.g., phenobarbital exposure). Precise developmental patterns have not been mapped out for most isoforms of cytochrome P450. For CYP1A2, studies using caffeine as a model substrate have revealed the pattern shown in Figure 34 (Lambert et al., 1986). Such a pattern has been noted for many compounds (e.g., theophylline, anticonvulsants) where a very limited metabolic clearance in the newborn matures during the first year of life (albeit with considerable intersubject and metabolic pathway variability) and ultimately achieves weight-adjusted clearance values that exceed those of adults. At puberty, clearance begins to decline, earlier in girls than in boys, to adult levels. The mechanisms regulating such developmental changes are uncertain, and other pathways of drug clearance likely mature with different patterns (deWildt et al., 1999). The critical point is that, at times of physiological change (the premature, the neonate, puberty), major changes in pharmacokinetics are likely to occur, variability is likely to be greatest (both within the same patient over time and among patients), and dosing adjustment, often aided by therapeutic drug monitoring for drugs with narrow therapeutic indices, becomes critical to safe, effective therapeutics. The 7-day-old neonate may be very different pharmacokinetically from the same patient as a newborn, and doses that were appropriate for a 10-year-old on a weight-adjusted basis might well result in overdose for the same patient at age 14.
Pharmacodynamic differences between children and adults have led to unexpected outcomes of therapy and adverse effects. For example, while antihistamines and barbiturates generally sedate adults, these drugs cause many children to become 'hyperactive.' Of great concern are the effects of medications, particularly when used chronically, on physical and cognitive development. Chronic therapy with phenobarbital can have a significant effect on learning and behavior in children. Tetracyclines deposit in developing teeth, with resultant permanent staining. While children are at risk for all the side effects of chronic corticosteroid therapy seen in adults, such drugs will also stunt linear growth. Children, however, are not always at increased risk for adverse drug effects. For example, while young children appear to be at higher risk for hepatotoxicity from valproic acid than are adults, they are at much lower risk for hepatotoxicity from isoniazid and possibly acetaminophen overdose. In 1997 Congress passed the Food and Drug Administration Modernization
Act (FDAMA). One of this act's aims is to enhance the amount of available
information on drug use in children. FDAMA and the Final Rule that followed
give the FDA the ability to request information on marketed drugs and reward
pharmaceutical companies with 6 months of additional marketing exclusivity
for studies that adequately address the request. For drugs in development, a
plan to obtain data in children must be negotiated with the FDA prior to the
drug's approval for marketing. The data required for children depend on the
disease being studied. If the disease is similar in adults and children and
there is no known reason to suspect the drug to behave differently in
children, then the bulk of the adult efficacy data can be extrapolated to
children if similar drug exposure in children and adults can be assured.
Thus, pharmacokinetic data in children are required along with adequate
exposure for safety assessment, but the FDA efficacy standard for adequate
and well-controlled trials prior to approval of the drug for children may not
be required. This approach by the FDA is novel and has already resulted in
the accumulation of much new data in children. The rest of the world has its
eyes on this initiative, and discussions are under way in Pediatric formulations of old and new drugs remain a problem for practical therapeutics. While toxicity of the vehicles used to administer drugs (e.g., diethylene glycol toxicity from elixir of sulfanilamide) led to the Pure Food and Drug Act of 1938, the 'gasping syndrome' associated with excess administration of drugs preserved with benzyl alcohol was described in the newborn as recently as the 1980s. For intravenous medications, formulations are often too concentrated for proper measurement of the tiny doses required for newborns. Oral formulations frequently present major problems with palatability and possible adverse reactions to flavoring and coloring agents. Particularly for pediatric suspensions, syrups, and chewable tablets, different preparations of the same drugs, while being equivalent from the point of view of bioavailability, may differ in acceptability to a specific patient. The Elderly As adults age, gradual changes in drug kinetics and effects result in an increase in the interindividual variability of doses required for a given effect. The pharmacokinetic changes result from changes in body composition and the function of drug-eliminating organs. The reduction in lean body mass, serum albumin, and total body water and the increase in percentage of body fat result in changes in the distribution of drugs depending on their lipid solubility and protein binding. The clearance of many drugs is reduced in the elderly. Renal function declines at a variable rate to about 50% of that in the young adult. Hepatic blood flow and the function of some of the drug-metabolizing enzymes also is reduced in the elderly, but the variability of this change is great. In general, the activities of cytochrome P450 enzymes are reduced, but conjugation mechanisms are relatively well maintained. Frequently, the elimination half-life of drugs is increased as a consequence of a larger apparent volume of distribution (of lipid-soluble drugs) and/or a reduction of the renal or metabolic clearance. Changes in pharmacodynamics also are important factors in treating the elderly. Drugs that depress the central nervous system produce increased effects at any given plasma concentration. Physiological changes and loss of homeostatic resilience can result in increased sensitivity to unwanted effects of drugs, such as hypotension from psychotropic medications and hemorrhage from anticoagulants, even if dosage is appropriately adjusted to account for the age-related pharmacokinetic changes. The proportion of our population in the elderly and very old age groups is increasing. These individuals have more illnesses than younger people and consume a disproportionate share of prescription and over-the-counter drugs. These factors, combined with the changes in pharmacokinetics and pharmacodynamics that occur with aging, make the elderly age group a population in whom drug use is likely to be marred by serious adverse drug effects and drug interactions. It is a population that should receive drugs only when absolutely necessary for well-defined indications and at the lowest effective doses. Prospectively defined endpoints, appropriate use of therapeutic drug monitoring, and frequent reviews of the patient's drug historywith discontinuation of those drugs that did not achieve the endpoint desired or are no longer requiredwould greatly improve the health of the elderly population. On the other hand, appropriate therapy should not be withheld because of these concerns. Outcomes data with a number of drug interventions have proven that the elderly can benefit at least as much as, and often more than, the young in the treatment of chronic diseases such as hypertension and hypercholesterolemia (LaRosa et al., 1999). Furthermore the natural history of chronic diseases of the elderly, such as osteoporosis and prostate hyperplasia, can be halted or reversed by appropriate drug therapy. Gender Although there may be some pharmacokinetic or pharmacodynamic differences between the sexes, early drug development until recently has been performed exclusively in males because of FDA guidelines that prohibited the participation of women of childbearing potential. In the 1990s, the FDA readdressed the importance of including women in early clinical trials, and the previous guidelines were revised to allow the participation of women in all phases of drug development. It is expected that at the time of drug approval, the database will be sufficiently complete to allow a rational assessment of the pharmacokinetic, pharmacodynamic, and safety issues in each sex (Sherman et al., 1995; Harris et al., 1995). DrugDrug Interactions The use of several drugs often is essential to obtain a desired therapeutic objective or to treat coexisting diseases. Examples abound, and the choice of drugs to be employed concurrently can be based on sound pharmacological principles. In the treatment of hypertension, a single drug is effective in only a modest percentage of patients. In the treatment of heart failure, the concurrent use of a diuretic with a vasodilator and/or a cardiac glycoside often is essential to achieve an adequate cardiac output and to keep the patient free from edema. Multiple-drug therapy is the norm in cancer chemotherapy and for the treatment of certain infectious diseases. The goals in these cases usually are to improve therapeutic effectiveness and to delay the emergence of malignant cells or of microorganisms that are resistant to the effects of available drugs. When physicians use several drugs concurrently, they face the problem of knowing whether a specific combination in a given patient has the potential to result in an interaction, and if so, how to take advantage of the interaction if it leads to improvement in therapy or how to avoid the consequences of an interaction if they are adverse. A potential drug interaction refers to the possibility that one drug may alter the intensity of pharmacological effects of another drug given concurrently. The net result may be enhanced or diminished effects of one or both of the drugs or the appearance of a new effect that is not seen with either drug alone. The frequency of significant beneficial or adverse drug interactions is unknown. Surveys that include data obtained in vitro, in animals, and in case reports tend to predict a frequency of interactions that is higher than actually occurs. While such reports have contributed to skepticism about the overall importance of drug interactions, there are potential interactions of definite clinical importance, and the physician must be alert to the possibility of their occurrence. Estimates of the incidence of clinical drugdrug interactions range from 3% to 5% in patients taking a few drugs to 20% in patients who are receiving 10 to 20 drugs. Because most hospitalized patients receive at least six drugs, the scope of the problem clearly is significant. The recent successful treatment of AIDS with multiple drugs, including several that have potent effects to alter the activity of drug-metabolizing enzymes, has heightened the public awareness of drug interactions. Recognition of beneficial effects and recognition and prevention of adverse drug interactions require a thorough knowledge of the intended and possible effects of drugs that are prescribed, an inclination to attribute unusual events to drugs rather than to disease, and adequate observation of the patient. Automated monitoring of prescription orders in the hospital or outpatient pharmacy may decrease the physician's need to memorize potential interactions. Nevertheless, knowledge of likely mechanisms of drug interactions is the only way the clinician can be prepared to analyze new findings systematically. It is incumbent upon the physician to be familiar with the basic principles of drugdrug interactions in planning a therapeutic regimen. Such reactions are discussed for individual drugs throughout this textbook. Interactions may be either pharmacokinetic (alteration of the absorption, distribution, or elimination of one drug by another) or pharmacodynamic (e.g., interactions between agonists and antagonists at drug receptors). The most important adverse drugdrug interactions occur with drugs that have serious toxicity and a low therapeutic index, such that relatively small changes in drug level can have significant adverse consequences. Additionally, drugdrug interactions can be clinically important if the disease being controlled with the drug is serious or potentially fatal if undertreated. Pharmacokinetic DrugDrug Interactions Drugs may interact at any point during their absorption, distribution, metabolism, or excretion; the result may be an increase or decrease in the concentration of drug at the site of action. As individuals vary in their rates of disposition of any given drug, the magnitude of an interaction that alters pharmacokinetic parameters is not always predictable, but it can be very significant. The delivery of drug into the circulation may be altered by physicochemical interactions that occur prior to absorption. For example, drugs may interact in an intravenous solution to produce an insoluble precipitate that may or may not be obvious. In the gut, drugs may chelate with metal ions or adsorb to medicinal resins. Thus, Ca2+ and other metallic cations contained in antacids are chelated by tetracycline, and the complex is not absorbed. Cholestyramine adsorbs and inhibits the absorption of thyroxine, cardiac glycosides, warfarin, corticosteroids, and probably other drugs. The rate and sometimes the extent of absorption can be affected by drugs that alter gastric motility, but this is usually of little clinical consequence. Interactions within the gut may be indirect and complex. Antibiotics that alter the gastrointestinal flora can reduce the rate of bacterial synthesis of vitamin K such that the effect of oral anticoagulants, which compete with vitamin K, will be enhanced. If a drug is metabolized by the gastrointestinal microorganisms, antibiotic therapy may result in an increase in the absorption of the drug, as has been demonstrated for some patients receiving digoxin (Lindenbaum et al., 1981). Recently, it has become evident that a number of drugs are substrates for various promiscuous transport systems that are present in many cells. P-glycoprotein (PGP) is the best studied of these systems, but many other systems are being discovered, such as the family of organic anion transporter systems. PGP is present in intestinal cells, renal tubular cells, biliary canalicular cells, and cells making up the blood-brain barrier. In the gut, PGP pumps drug into the lumen and thereby limits absorption. In the blood-brain barrier, PGP eliminates drug from the central nervous system (CNS), thus altering drug distribution. In the liver and kidney, PGP transports drug into the biliary canalicula and tubular lumen, thereby enhancing drug elimination. Inhibition of PGP therefore can alter the absorption, distribution, and elimination of drugs and is a topic of much current investigation. Cyclosporin A, quinidine, verapamil, itraconazole, and clarithromycin are examples of drugs that can inhibit PGP, whereas rifampin apparently can induce PGP. It is curious that inhibitors and inducers of CYP3A4 often appear to have similar effects on PGP, although this is not always true (Kim et al., 1999). Much as there has been an explosion of information in the past decade about the CYP drug-metabolizing enzymes, the next decade promises a rich yield of information on PGP and similar transport systems. Many drugs are extensively bound to plasma albumin (acidic drugs) or A few drugs are actively transported to their site of action. For instance, the antihypertensive drugs guanethidine and guanadrel inhibit sympathetic nervous system function after being transported into adrenergic neurons by the norepinephrine-uptake mechanism. Inhibition of this neuronal uptake system by tricyclic antidepressants and some sympathomimetic amines will inhibit the sympathetic blockade and reduce the antihypertensive effects of guanethidine and guanadrel. More drugs may be transported away from their site of action by PGP or other transporters. For example, cancer chemotherapy may be limited by transport of anticancer drugs out of tumor cells by PGP. Attempts have been made to block PGP in order to enhance chemotherapy, thus making use of a drugdrug interaction to enhance clinical efficacy (Krishan et al., 1997). Interactions involving drug metabolism can increase or decrease the
amount of drug available for action by inhibition or induction of metabolism,
respectively (see also Chapter 1: Pharmacokinetics: The Dynamics of
Drug Absorption, Distribution, and Elimination). Interactions may occur among
administered drugs or between drugs and dietary substances [e.g.,
grapefruit juice (a CYP3A4 inhibitor)], herbal remedies [e.g., The ability of one drug to inhibit the renal excretion of another is dependent on an interaction at active transport sites. Many of the reported interactions occur at the anion transport site, where, for example, probenecid inhibits the excretion of penicillin to cause the desirable effects of elevated plasma concentrations of the antibiotic and a longer half-life. Similarly, the renal elimination of methotrexate is inhibited by probenecid, salicylates, and phenylbutazone, but in this case methotrexate toxicity may result from the interaction. Interactions at the transport site for basic drugs include the inhibition of excretion of procainamide by cimetidine and amiodarone. An interaction at renal tubular PGP causes inhibition of the excretion of digoxin by quinidine, verapamil, and amiodarone. Finally, the excretion of Li+ can be affected by drugs that alter the ability of the proximal renal tubule to reabsorb Na+. Thus, clearance of Li+ is reduced and concentrations of Li+ in plasma are increased by diuretics that cause volume depletion and by nonsteroidal antiinflammatory drugs that enhance proximal tubular reabsorption of Na+. Pharmacodynamic DrugDrug Interactions There are numerous examples of drugs that interact at a common
receptor site or that have additive or inhibitory effects due to actions at
different sites in an organ. Such interactions are described throughout this
textbook. Frequently overlooked is the multiplicity of effects of many drugs.
Thus, phenothiazines are effective Other interactions of an apparently pharmacodynamic nature are poorly understood or are mediated indirectly. Halogenated hydrocarbons, including many general anesthetics, sensitize the myocardium to the arrhythmogenic actions of catecholamines. This effect may result from an action on the pathway that leads from adrenergic receptor to effector, but the details are unclear. The striking interaction between meperidine and monoamine oxidase inhibitors to produce seizures and hyperpyrexia may be related to excessive amounts of an excitatory neurotransmitter, but the mechanism has not been elucidated. One drug may alter the normal internal milieu, thereby augmenting or diminishing the effect of another agent. A well-known example of such an interaction is the enhancement of the toxic effects of digoxin as a result of diuretic-induced hypokalemia. Summary: DrugDrug Interactions Drugdrug interactions are only one of the many factors discussed in this chapter that can alter the patient's response to therapy. The major task of the physician is to determine if an interaction has occurred and the magnitude of its effect. When unexpected effects are seen, a drug interaction should be suspected. Careful drug histories are important, because patients may take over-the-counter drugs or herbal products, take drugs prescribed by another physician, or take drugs prescribed for another patient. Care must be exercised when major changes are made in a drug regimen, and drugs that are not necessary should be discontinued. When an interaction is discovered, the interacting drugs often may be used effectively with adjustment of dosage or other therapeutic modifications. Fixed-Dose Combinations The concomitant use of two or more drugs adds to the complexity of individualization of drug therapy. The dose of each drug should be adjusted to achieve optimal benefit. Thus, patient compliance is essential yet more difficult to achieve. To obviate the latter problem, many fixed-dose drug combinations are marketed. The use of such combinations is advantageous only if the ratio of the fixed doses corresponds to the needs of the individual patient. In the Placebo Effects The net effect of drug therapy is the sum of the pharmacological effects of the drug and the nonspecific placebo effects associated with the therapeutic effort. Although identified specifically with administration of an inert substance in the guise of medication, placebo effects are associated with the taking of any drug, active or inert. Placebo effects result presumably from the physicianpatient relationship, the significance of the therapeutic effort to the patient, or the mental set imparted by the therapeutic setting and by the physician. They vary significantly in different individuals and in any one patient at different times. Placebo effects commonly are manifested as alterations of mood, other subjective effects, and objective effects that are under autonomic or voluntary control. They may be favorable or unfavorable relative to the therapeutic objectives. Exploited to advantage, placebo effects can significantly supplement pharmacological effects and can represent the difference between success and failure of therapy. A placebo (in this context, better termed dummy medication) is an indispensable element of many controlled clinical trials. In contrast, a placebo has only a limited role in the routine practice of medicine. A supportive physicianpatient relationship generally is preferable to the use of a placebo for promoting therapeutic benefits. Relief or lack of relief of symptoms upon administration of a placebo is not a reliable basis for determining whether the symptoms have a 'psychogenic' or 'somatic' origin. Tolerance Tolerance may be acquired to the effects of many drugs, especially the opioids, various CNS depressants, and organic nitrates. When this occurs, cross-tolerance may develop to the effects of pharmacologically related drugs, particularly those acting at the same receptor site, and drug dosage must be increased to maintain a given therapeutic effect. Since tolerance does not usually develop equally to all effects of a drug, the therapeutic index may decrease. However, there also are examples of the development of tolerance to the undesired effects of a drug and a resultant increase in its therapeutic index (e.g., tolerance to sedation produced by phenobarbital when used as an anticonvulsant). The mechanisms involved in the development of tolerance are only partially understood. Tolerance may occur as the result of induced synthesis of the hepatic microsomal enzymes involved in drug biotransformation. Another example of pharmacokinetic tolerance is the development of resistance of cancer cells to drug-induced cytotoxicity due to the induction of PGP, which transports drug out of the cell, thereby reducing the intracellular concentration of the chemotherapeutic agent. The most important factor in the development of tolerance to the opioids, barbiturates, ethanol, and organic nitrates is a type of cellular adaptation referred to as pharmacodynamic tolerance; multiple mechanisms are involved, including changes in the number, affinity, or function of drug receptors. Tachyphylaxis, such as that to histamine-releasing agents and to the sympathomimetic amines that act indirectly by releasing norepinephrine, has been attributed to depletion of available mediator, but other mechanisms also may contribute. The subject of tolerance is discussed in more detail in Chapter 24: Drug Addiction and Drug Abuse. Genetic Factors Genetic factors are the major determinants of the normal variability of drug effects and are responsible for a number of striking quantitative and qualitative differences in pharmacological activity. Basic principles of human genetics apply to genetic loci coding for proteins involved in handling of drugs, e.g., drug metabolizing enzymes, carrier proteins, and receptors. Thus (1) allelic variation is common; (2) there are often several different alleles producing variant proteins at a given locus; (3) some allelic variants are 'silent,' with no functional consequences, while others may markedly alter the handling of foreign compounds; (4) gene frequencies for different alleles are likely to vary among different human populations, suggesting the need for vigilance in extrapolation of kinetic and safety data from one population to another; (5) some allelic variants are classified as 'polymorphisms,' variant alleles with a frequency of at least 1%, while other, less common variants are classified as 'rare inborn errors of metabolism.' The consequences of pharmacogenetic variation include: (1) altered clearance of drugs, resulting in a 'functional overdose' in those individuals unable to metabolize the compound; (2) failure to convert a prodrug to an active drug; (3) altered pharmacodynamics (e.g., hemolytic anemia secondary to glucose-6-phosphate dehydrogenase deficiency); and (4) idiosyncratic drug reactions, such as aplastic anemia or hepatotoxicity. The superfamily of cytochrome P450 enzymes has been extensively investigated for pharmacogenetic variants (Ingelman-Sundberg et al., 1999). For example, an abnormality in CYP2D6 (present in 3% to 10% of various populations) results in deficient metabolism of many compounds. For some of these drugsfor example, the tricyclic antidepressantstoxicity of 'standard' doses may result from accumulation when used in CYP2D6-deficient patients, while for other drugs, either because of a wide therapeutic index (e.g., dextromethorphan) or because multiple pathways are involved in clearance (e.g., propranolol), no dosage adjustment is required. During drug development, compounds may be screened in vitro with human tissue preparations or recombinantly expressed human cytochrome P450 enzymes to ascertain if pharmacogenetic polymorphisms are likely to be involved in metabolism of the drug. Single-dose studies in subjects genotyped for various polymorphisms may help clarify whether the potential for altered drug handling is clinically relevant. For pharmacogenetics to become clinically useful, molecular diagnostic tests for pharmacogenetic variants, done in routine clinical laboratories, must become available, so that a physician can individualize choice of medication or dose regimen based on each specific patient's drug metabolism profile. If a relatively rare but severe adverse reaction to a drug (e.g., a 1 in 5000 risk of hepatotoxicity) is strongly linked to a given pharmacogenetic polymorphism, such pharmacogenetic 'prescreens' could markedly decrease the risk for individual patients and the population as a whole. Approach to Individualization After it has been determined that pharmacotherapy is necessary to modify the symptoms or outcome of a disease, the therapist is faced with two types of decisions: the first is qualitative (the initial choice of a specific drug) and the second is quantitative (the initial dosage regimen). Optimal treatment will result only when the physician is aware of the sources of variation in response to drugs and when the dosage regimen is designed on the basis of the best available data about the diagnosis, severity, and stage of the disease, presence of concurrent diseases or drug treatment, and predefined goals of acceptable efficacy and limits of acceptable toxicity. If objectively assessable expectations of drug therapy are not set before therapy is initiated, therapy is likely to be ineffective and continued longer than necessary unless an obvious adverse effect occurs. In most clinical settings, the decision about the choice of drug is influenced substantially by the confidence the physician has in the accuracy of the diagnosis and estimates of the extent and severity of disease. Based on the best available information, the physician must decide on an initial drug from a group of reasonable alternatives. The extent of this evaluation is itself dependent on many factors, including a cost-benefit analysis of diagnostic tests, and this must be based on the availability and specificity of alternative therapies and the likelihood of a reduction in future utilization of expensive health care. The initial dosage regimen is determined by estimation, if possible, of the pharmacokinetic properties of the drug in the individual patient. The estimate must be based on an appreciation of the variables that are most likely to affect the disposition of the particular drug. These variables have been discussed above (see Figure 31 and Appendix II). Subsequent adjustments may be aided in some instances by measurement of drug concentrations but must ultimately be based on whether the regimen is efficacious, either without adverse effects or at an acceptable level of toxicity. It has been stated above that every therapeutic plan is and should be treated as an experiment. As such, most of the considerations that were specified in the discussion of clinical trials must be applied to individual patients. Of utmost importance is the definition of specific goals of treatment and the means to assess whether or not these goals are being achieved. Whenever possible, the objective endpoint should be related as closely as possible to the clinical goals of therapy (e.g., shrinkage of a tumor or eradication of an infection). Many clinical goals are, however, difficult to assess (e.g., the prevention of cardiovascular complications associated with hypertension and diabetes). In such cases, it is necessary to use surrogate markers, such as a reduction in blood pressure or the concentration of glucose or cholesterol in plasma. These intermediate endpoints are based on demonstrated (in clinical trials) or assumed correlation of the surrogate marker with the ultimate clinical benefit. In many casessuch as improvement of exercise tolerance in patients with congestive heart failure, the elimination of asymptomatic ventricular arrhythmias, or the change in CD4 lymphocyte count in AIDSthe link between the surrogate marker and the ultimate goal is controversial (Fleming and DeMets, 1996). The value or utility of each regimen must be assessed at intervals during the course of therapy. The utility of a regimen can be defined as the benefit it produces plus the dangers of not treating the disease minus the sum of the adverse effects of therapy. Another common expression of the usefulness of a regimen is its ratio of risks to benefits (representing a balance between the efficacious and toxic effects of the drug). A definitive evaluation of the utility of a drug is not easy; nevertheless, some sense of the value of a regimen must be established in the minds of the physician and the patient. Knowledge of the usefulness of a given regimen may be a critical determinant of protracted compliance by the patient to a long-term regimen or logical discontinuation by the physician of a marginally efficacious and risky therapy. It must be remembered that the physician, the patient, and the patient's family may have disparate opinions of the utility of a therapeutic regimen. In one study of antihypertensive therapy in which all patients were judged to be improved by the physician, only 48% of the patients considered themselves improved and 8% felt worse. Relatives thought that only 1% of the patients were improved and that 99% had evidence of adverse effects of therapy (Jachuck et al., 1982). |
Drug Regulation and Development
|
Drug Regulation The history of drug regulation in the In this relatively relaxed atmosphere, research in basic and clinical
pharmacology burgeoned in both industrial and academic laboratories. The
result was a flow of new drugs, called 'wonder drugs' by the lay
press, for the treatment of both infectious and organic disease. Because
efficacy was not rigorously defined, a number of therapeutic claims could not
be supported by data. The risk-to-benefit ratio was seldom mentioned, but it
emerged in dramatic fashion early in the 1960s. At that time, thalidomide, a
hypnotic with no obvious advantage over other drugs in its class, was
introduced in The Harris-Kefauver Amendments are sound legislation. They require
sufficient pharmacological and toxicological research in animals before a
drug can be tested in human beings. The data from such studies must be
submitted to the FDA in the form of an application for an investigational new
drug ( To demonstrate efficacy, 'adequate and well-controlled investigations' must be performed. This generally has been interpreted to mean two replicate clinical trials that are usually, but not always, randomized, double blind, and placebo controlled. Safety is demonstrated by having a sufficiently large database of patients/subjects who have received the drug at the time of filing an NDA with the FDA for approval. As a result of these requirements, the number of patients on the drug, the number of studies, the development cost, and the time required for the clinical studies to complete the NDA have increased. The regulatory review time also increased as a result of the mass and complexity of the data, so that by 1990 the average review time was approaching three years. This increased the inherent tension that exists between the FDA, which is motivated to protect the public health, and the drug developers, who are motivated to market effective and profitable drugs. Competing pressures also exist in the community, where medical practitioners and patient activist groups have criticized the FDA for delaying approval, while some 'watchdog' groups criticize the FDA for allowing drugs on the market that occasionally cause unexpected problems after they are marketed. The FDA has the difficult task of balancing the requirement that drugs be safe and effective and yet allowing useful medications to be made available in a timely manner. Beginning in the late 1980s with pressure from AIDS activists, the FDA
undertook a number of initiatives that have had profound effects in
streamlining the process of regulatory approval. These initiatives have all
but eliminated the concern about the 'drug lag,' where drugs were
available in other countries significantly sooner than in the
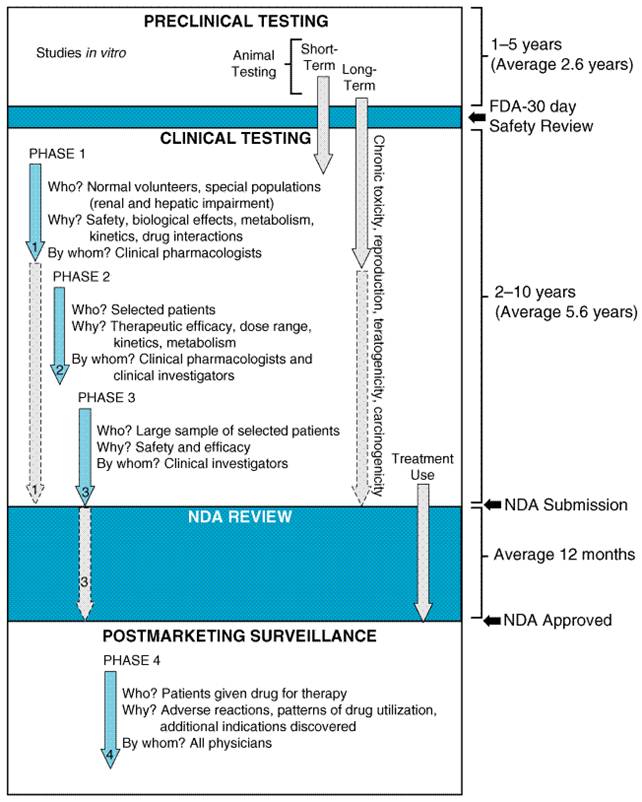
A seemingly contradictory directive to the FDA also is contained in the Food, Drug, and Cosmetic Actthat is, the FDA cannot interfere with the practice of medicine. Thus, once the efficacy of a new agent has been proven in the context of acceptable toxicity, the drug can be marketed. The physician then is allowed to determine its most appropriate use. However, physicians must realize that new drugs are inherently more risky because of the relatively small amount of data about their effects. Yet there is no practical way to increase knowledge about a drug before it is marketed. A systematic method for postmarketing surveillance is an indispensable requirement for early optimization of drug use. Before a drug can be marketed, a package insert for use by physicians must be prepared. This is a cooperative effort between the FDA and the pharmaceutical company. The insert usually contains basic pharmacological information as well as essential clinical information in regard to approved indications, contraindications, precautions, warnings, adverse reactions, usual dosage, and available preparations. Promotional materials cannot deviate from information contained in the insert. One area in which the FDA does not have clear authority is in the regulation of 'dietary supplements,' including vitamins, minerals, proteins, and herbal preparations. Until 1994, the FDA regulated such supplements as either food additives or drugs, depending on the substance and the indications that were claimed. However, in 1994, Congress passed the Dietary Supplement Health and Education Act (DSHEA), which weakened the authority of the FDA. The Act defined dietary supplement as a product intended to supplement the diet that contains '(A) a vitamin; (B) a mineral; (C) an herb or other botanical; (D) an amino acid; (E) a dietary substance for use by man to supplement the diet by increasing the total daily intake; or (F) a concentrate, metabolite, constituent, extract or combination of an ingredient described in clause (A), (B), (C), (D), or (E).' Such products must be labeled as 'dietary supplement.' The FDA does not have the authority to require approval prior to marketing of such supplements unless the supplements make specific claims relating to the diagnosis, treatment, prevention, or cure of a disease. However, the common conditions associated with natural statessuch as pregnancy, menopause, aging, and adolescencewill not be treated as diseases by the FDA. Treatment of hot flashes, symptoms of the menstrual cycle, morning sickness associated with pregnancy, mild memory problems associated with aging, hair loss, and noncystic acne are examples of claims that can be made without prior FDA approval. Also, health maintenance and other 'nondisease' claims such as 'helps you relax' or 'maintains a healthy circulation' are allowed without approval. Many supplements with such claims are labeled as follows: 'This statement has not been evaluated by the FDA. This product is not intended to diagnose, treat, cure, or prevent any disease.' The FDA cannot remove such products from the market unless they can prove that there is a 'significant or unreasonable risk of illness or injury' when the product is used as directed or under normal conditions of use. It is the manufacturer's responsibility to ensure that its products are safe. As a result of the DSHEA legislation, a large number of unregulated products that have not been demonstrated to be safe or effective are widely available. There have been several occasions where such products have been associated with serious adverse effects or have been shown to interact with prescription drugs (see Fugh-Berman, 2000). Under these circumstances, the FDA can act, but the burden is on the FDA to prove the supplements are unsafe. In many ways this situation is analogous to the lack of regulation of drugs that existed prior to the 1938 disaster involving elixir of sulfanilamide, described above. Physicians and patients alike should be aware of the lack of regulation of dietary supplements. Adverse reactions or suspected interactions with such substances should be reported to the FDA using the same mechanisms as for adverse drug reactions (see Adverse Drug Reactions and Drug Toxicity). Drug Development Except for concern about governmental interference with the practice of medicine, the average physician has not considered it important to understand the process of drug development. Yet, an appreciation of this process is necessary to estimate the risk-to-benefit ratio of a drug and to realize the limitations of the data that support the efficacy and safety of a marketed product. By the time an Trials of drugs in human beings in the |
Adverse Drug Reactions and Drug Toxicity
|
Any drug, no matter how trivial its therapeutic actions, has the potential to do harm. Adverse reactions are a cost of modern medical therapy. Although the mandate of the FDA is to ensure that drugs are safe and effective, both of these terms are relative. The anticipated benefit from any therapeutic decision must be balanced by the potential risks. Patients, to a greater extent than physicians, are unaware of the limitations of the premarketing phase of drug development in defining even relatively common risks of new drugs. Since only a few thousand patients are exposed to experimental drugs in more or less controlled and well-defined circumstances during drug development, adverse drug effects that occur as frequently as 1 in 1000 patients may not be detected prior to marketing. Postmarketing surveillance of drug usage is thus imperative to detect infrequent but significant adverse effects. 'Mechanism-based' adverse drug reactions (extensions of the principal pharmacological action of the drug) are relatively easily predicted by preclinical and clinical pharmacology studies. For 'idiosyncratic' adverse reactions, which result from an interaction of the drug with unique host factors that are unrelated to the principal action of the drug, current approaches to 'safety assessment,' both preclinically and in clinical trials, are problematic. The relative rarity of severe idiosyncratic reactions (e.g., severe dermatological, hematological, or hepatological toxicities) presents epidemiological ascertainment issues. In addition, it is clear that a population risk of 1 in 1000 is not distributed evenly across the population; some patients, because of unique genetic or environmental factors, are at an extremely high risk, while the remainder of the population may be at low or no risk. In contrast to the human heterogeneity underlying idiosyncratic risk, the standard process of drug developmentparticularly the preclinical safety assessment using inbred healthy animals maintained in a defined environment on a defined diet and manifesting predictable habitslimits the identification of risk for idiosyncratic adverse drug reactions in the human population. Understanding the genetic and environmental bases of idiosyncratic adverse events holds the promise of assessing individual rather than population risk, thereby improving the overall safety of pharmacotherapy. Postmarketing Detection of Adverse Reactions Several strategies exist to detect adverse reactions after marketing of a drug, but debate continues about the most efficient and effective method. Formal approaches for estimation of the magnitude of an adverse drug effect are the follow-up or 'cohort' study of patients who are receiving a particular drug, the 'case-control' study, where the potential for a drug to cause a particular disease is assessed, and meta-analysis of pre- and postmarketing studies. Cohort studies can estimate the incidence of an adverse reaction, but they cannot, for practical reasons, discover rare events. To have any significant advantage over the premarketing studies, a cohort study must follow at least 10,000 patients who are receiving the drug to detect with 95% confidence one event that occurs at a rate of 1 in 3300, and the event can be attributed to the drug only if it does not occur spontaneously in the control population. If the adverse event occurs spontaneously in the control population, substantially more patients and controls must be followed to establish the drug as the cause of the event (Strom and Tugwell, 1990). Meta-analyses combine the data from several studies in an attempt to discern benefits or risks that are sufficiently uncommon that an individual study lacks the power to discover them (Temple, 1999). Case-control studies also can discover rare drug-induced events. However, it may be difficult to establish the appropriate control group (Feinstein and Horwitz, 1988), and a case-control study cannot establish the incidence of an adverse drug effect. Furthermore, the suspicion of a drug as a causative factor in a disease must be the impetus for the initiation of such case-control studies. The magnitude of the problem of adverse reactions to marketed drugs is
difficult to quantify. It has been estimated that 3% to 5% of all
hospitalizations can be attributed to adverse drug reactions, resulting in
300,000 hospitalizations annually in the Because of the shortcomings of cohort and case-control studies and
meta-analyses, other approaches must be used. Spontaneous reporting of
adverse reactions has proven to be an effective way to generate an early
signal that a drug may be causing an adverse event. It is the only practical
way to detect rare events, events that occur after prolonged use of drug,
adverse effects that are delayed in appearance, and many drugdrug
interactions. In the past few years, considerable effort has gone into
improving the reporting system in the The most important spontaneous reports are those that describe serious reactions, whether they have been described previously or not. Reports on newly marketed drugs (within the past 3 years) are the most significant, even though the physician may not be able to attribute a causal role to a particular drug. The major use of this system is to provide early warning signals of unexpected adverse effects that can then be investigated by more formal techniques. However, the system also serves to monitor changes in the nature or frequency of adverse drug reactions due to aging of the population, changes in the disease itself, or the introduction of new, concurrent therapies. The primary sources for the reports are responsible, alert physicians; other potentially useful sources are nurses, pharmacists, and students in these disciplines. In addition, hospital-based pharmacy and therapeutics committees and quality assurance committees frequently are charged with monitoring adverse drug reactions in hospitalized patients, and reports from these committees should be forwarded to the FDA. The simple forms for reporting may be obtained 24 hours a day, 7 days a week by calling (800)-FDA-1088, or reporting can be done directly on the Internet (https://www.fda.gov/medwatch). Additionally, health professionals may contact the pharmaceutical manufacturer, who is legally obligated to file reports with the FDA. |
Guide to the 'Therapeutic Jungle'
|
The flood of new drugs in recent years has provided many dramatic improvements in therapy, but it also has created a number of problems of equal magnitude. Not the least of these is the 'therapeutic jungle,' the term used to refer to the combination of the overwhelming number of drugs, the confusion over nomenclature, and the associated uncertainty of the status of many of these drugs. A reduction in the marketing of close congeners and drug mixtures and an improvement in the quality of advertising are important ingredients in the remedy for the therapeutic jungle. However, physicians also can contribute to the remedy by employing nonproprietary rather than proprietary names whenever appropriate, by using prototypes both as an instructional device and in clinical practice, by adopting a properly critical attitude toward new drugs, and by knowing and making use of reliable sources of pharmacological information. Most important, they should develop a 'way of thinking about drugs' based upon pharmacological principles. Drug Nomenclature The existence of many names for each drug, even when the names are reduced to a minimum, has led to a lamentable and confusing situation in drug nomenclature. In addition to its formal chemical name, a new drug is usually assigned a code name by the pharmaceutical manufacturer. If the drug appears promising and the manufacturer wishes to place it on the market, a United States Adopted Name (USAN) is selected by the USAN Council, which is jointly sponsored by the American Medical Association, the American Pharmaceutical Association, and the United States Pharmacopeial Convention, Inc. This nonproprietary name often is referred to as the generic name. If the drug is eventually admitted to the United States Pharmacopeia (see below), the USAN becomes the official name. However, the nonproprietary and official names of an older drug may differ. Subsequently, the drug also will be assigned a proprietary name or trademark by the manufacturer. If the drug is marketed by more than one company, it may have several proprietary names. If mixtures of the drug with other agents are marketed, each such mixture also may have a separate proprietary name. There is increasing worldwide adoption of the same name for each therapeutic substance. For newer drugs, the USAN is usually adopted for the nonproprietary name in other countries, but this is not true for older drugs. International agreement on drug names is mediated through the World Health Organization and the pertinent health agencies of the cooperating countries. One area of continued confusion and ambiguity is the designation of the stereochemical composition in the name of a drug. The nonproprietary names usually give no indication of the drug's stereochemistry except for a few drugs such as levodopa and dextroamphetamine. Even the chemical names cited by the USAN Council often are ambiguous. Physicians and other medical scientists are frequently ignorant about drug stereoisomerism and are likely to remain so until the system of nonproprietary nomenclature incorporates stereoisomeric information (Gal, 1988). The nonproprietary or official name of a drug should be used whenever possible, and such a practice has been adopted in this textbook. The use of the nonproprietary name is clearly less confusing when the drug is available under multiple proprietary names and when the nonproprietary name more readily identifies the drug with its pharmacological class. The best argument for the proprietary name is that it is frequently more easily pronounced and remembered as a result of advertising. For purposes of identification, representative proprietary names, designated by SMALLCAP TYPE, appear throughout the text as well as in the index. Not all proprietary names for drugs are included, because the number of proprietary names for a single drug may be large and because proprietary names differ from country to country. The Drug Price Competition and Patent Term Restoration Act of 1984 allows more generic versions of brand-name drugs to be approved for marketing. When the physician prescribes drugs, the question arises as to whether the nonproprietary name or a proprietary name should be employed. A pharmacist may substitute a preparation that is equivalent unless the physician indicates 'no substitution' or specifies the manufacturer on the prescription. In view of the discussion above on the individualization of drug therapy, it is understandable why a physician who has carefully adjusted the dose of a drug to a patient's individual requirements for chronic therapy may be reluctant to surrender control over the source of the drug that the patient receives (Burns, 1999). Based on a number of considerationssuch as the frequency of use of a drug that is available from only a single manufacturer, the cost of filling a prescription, and the markup of the pharmacistit appears as though the overall savings to society of prescribing the least expensive nonproprietary preparation is about 5% (see Trout and Lee, 1981). Of course, savings in individual situations can be very much greater. On the other hand, the lower wholesale cost of the nonproprietary preparation sometimes is not passed on to the consumer (Bloom et al., 1986). More importantly, prescribing by nonproprietary name could result in the patient receiving a preparation of inferior quality or of uncertain bioavailability, and therapeutic failures due to decreased bioavailability have been reported (Hendeles et al., 1993). To address this issue, the FDA has established standards for bioavailability and compiled information about the interchangeability of drug products, which are published annually (Approved Drug Products with Therapeutic Equivalence Evaluations). Because of potential cost savings to the individual patient and simplification of the 'therapeutic jungle,' nonproprietary names should be used when prescribing except for drugs with a low therapeutic index and known differences in bioavailability among marketed products (Hendeles et al., 1993). Use of Prototypes It is obviously crucial for the physician to be thoroughly familiar with the pharmacological properties of a drug before it is administered. It follows that the patient will benefit if the physician avoids the temptation to choose from many different drugs for the patient's regimen. A physician's needs for therapeutic agents usually can be satisfied by thorough knowledge of one or two drugs in each therapeutic category. Inevitably, a small number of drugs can be used more effectively. When the clinical setting calls for a drug that the physician uses infrequently, he or she should feel obligated to learn about its effects, to use great caution in its administration, and to apply appropriate procedures in monitoring its effects. For teaching purposes in this textbook, the confusion created by the welter of similar drugs is reduced by restricting major attention to prototypes in each pharmacological class. A teaching prototype is often the agent most likely to be employed in clinical use, but this is not always true. A particular drug may be retained as the prototype, even though a new congener is clinically superior, either because more is known about the older drug or because it is more illustrative for the entire class of agents. Attitude Toward New Drugs A reasonable attitude toward new drugs is summarized by the adage that advises the physician to be 'neither the first to use a new drug nor the last to discard the old.' Only a minor fraction of new drugs represents a significant therapeutic advance. The limitation of information about toxicity and efficacy at the time of release of a drug has been emphasized above, and this is particularly pertinent to comparisons with older agents in the same therapeutic class. Nevertheless, the important advances in therapeutics in the last fifty years emphasize the obligation to keep abreast of significant advances in pharmacotherapy. |
Sources of Drug Information
|
The physician's need for objective, concise, and well-organized information on drugs is obvious. Among the available sources are textbooks of pharmacology and therapeutics, leading medical journals, drug compendia, professional seminars and meetings, and advertising. Despite this cornucopia of information, responsible medical spokespeople insist that most practicing physicians are unable to extract the objective and unbiased data required for the practice of rational therapeutics (Woosley, 1994). Depending on their aim and scope, pharmacology textbooks provide (in varying proportions) basic pharmacological principles, critical appraisal of useful categories of therapeutic agents, and detailed descriptions of individual drugs or prototypes that serve as standards of reference for assessing new drugs. In addition, pharmacodynamics and pathological physiology are correlated. Therapeutics is considered in virtually all textbooks of medicine, but often superficially. The source of information described as most often used by physicians in an industry survey is the Physicians' Desk Reference (PDR). The brand-name manufacturers whose products appear support this book. No comparative data on efficacy, safety, or cost are included. The information is identical to that contained in drug package inserts, which are largely based on the results of phase 3 testing; its primary value is thus in learning what indications for use of a drug have been approved by the FDA. There are, however, several inexpensive, unbiased sources of information on the clinical uses of drugs that provide balanced comparative data. All recognize that the physician's legitimate use of a drug in a particular patient is not limited by FDA-approved labeling in the package insert. The United States Pharmacopeia Dispensing Information (USPDI), first published in 1980, comes in two volumes. One, Drug Information for the Health Care Professional, consists of drug monographs that contain practical, clinically significant information aimed at minimizing the risks and enhancing the benefits of drugs. Monographs are developed by USP staff and are reviewed by advisory panels and other reviewers. The Advice for the Patient volume is intended to reinforce, in lay language, the oral consultation provided by the therapist, and this may be provided to the patient in written form. These volumes are published frequently. AMA Drug Evaluations, compiled by the American Medical Association Department of Drugs in cooperation with the American Society for Clinical Pharmacology and Therapeutics, includes general information on the use of drugs in special settings (e.g., pediatrics, geriatrics, renal insufficiency, etc.) and reflects the consensus of a panel on the effective clinical use of therapeutic agents. Facts and Comparisons also is organized by pharmacological classes and is updated monthly. Information in monographs is presented in a standard format and incorporates FDA-approved information, which is supplemented with current data obtained from the biomedical literature. A useful feature is the comprehensive list of preparations with a 'Cost Index,' an index of the average wholesale price for equivalent quantities of similar or identical drugs. Many of these publications are available on diskette or CD-ROM for personal computers. Industry promotionin the form of direct-mail brochures, journal advertising, displays, professional courtesies, or the detail person or pharmaceutical representativeis intended to be persuasive rather than educational. The pharmaceutical industry cannot, should not, and indeed does not purport to be responsible for the education of physicians in the use of drugs. Over 1500 medical journals are published regularly in the The United States Pharmacopeia (USP) and National Formulary
(NF) were recognized as 'official compendia' by the Federal Food
and Drug Act of 1906. The approved therapeutic agents used in medical
practice in the |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1353
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved