| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Drugs Effective in the Therapy of the Epilepsies
Overview
|
The epilepsies are common and frequently
devastating disorders, affecting approximately 2.5 million people in the The mechanisms of action of antiseizure drugs fall into three major categories. Drugs effective against the most common forms of epileptic seizures, partial and secondarily generalized tonic-clonic seizures, appear to work by one of two mechanisms. One is to limit the sustained, repetitive firing of a neuron, an effect mediated by promoting the inactivated state of voltage-activated Na+ channels. A second mechanism appears to involve enhanced gamma-aminobutyric acid (GABA)mediated synaptic inhibition, an effect mediated by an action presynaptically for some drugs and postsynaptically for others. Drugs effective against a less common form of epileptic seizure, absence seizure, limit activation of a particular voltage-activated Ca2+ channel known as the T current. Although many treatments are available, much effort is being devoted to novel approaches. Many of these approaches center on elucidating the genetic, cellular, and molecular mechanisms of the hyperexcitability, insights that promise to provide specific targets for novel therapies. |
Terminology and Epileptic Seizure Classification
|
The term seizure refers to a transient alteration of behavior due to the disordered, synchronous, and rhythmic firing of populations of brain neurons. The term epilepsy refers to a disorder of brain function characterized by the periodic and unpredictable occurrence of seizures. Seizures can be 'nonepileptic' when evoked in a normal brain by treatments such as electroshock or chemical convulsants or 'epileptic' when occurring without evident provocation. Pharmacological agents in current clinical use inhibit seizures, and thus are referred to as antiseizure drugs. Whether any of these agents has prophylactic value in preventing development of epilepsy (epileptogenesis) is uncertain. Seizures are thought to arise from the cerebral cortex, and not from other central nervous system (CNS) structures such as thalamus, brainstem, or cerebellum. Epileptic seizures have been classified into partial seizures, those beginning focally in a cortical site, and generalized seizures, those that involve both hemispheres widely from the outset (Commission, 1981). The behavioral manifestations of a seizure are determined by the functions normally served by the cortical site at which the seizure arises. For example, a seizure involving motor cortex is associated with clonic jerking of the body part controlled by this region of cortex. A simple partial seizure is associated with preservation of consciousness. A complex partial seizure is associated with impairment of consciousness. The majority of complex partial seizures originate from the temporal lobe. Examples of generalized seizures include absence, myoclonic, and tonic-clonic. The type of epileptic seizure determines the drug selected for therapy. More detailed information is presented in Table 211. Apart from this epileptic seizure classification, an additional classification specifies epileptic syndromes, which refer to a cluster of symptoms frequently occurring together and include seizure types, etiology, age of onset, and other factors (Commission, 1989). More than 40 distinct epileptic syndromes have been identified. The epileptic syndromes have been categorized into partial versus generalized epilepsies. The partial epilepsies may consist of any of the partial seizure types (Table 211) and account for roughly 60% of all epilepsies. The etiology commonly consists of a lesion in some part of the cortex, such as a tumor, developmental malformation, damage due to trauma or stroke, etc. Such lesions often are evident on brain imaging studies such as magnetic resonance imaging. Alternatively, the etiology may be genetic. The generalized epilepsies are characterized most commonly by one or more of the generalized seizure types listed in Table 211 and account for approximately 40% of all epilepsies. The etiology is usually genetic. The most common generalized epilepsy is referred to as juvenile myoclonic epilepsy, accounting for approximately 10% of all epileptic syndromes. The age of onset is in the early teens, and the condition is characterized typically by myoclonic and tonic-clonic and often absence seizures. Like most of the generalized-onset epilepsies, juvenile myoclonic epilepsy is a complex genetic disorder that is probably due to inheritance of multiple susceptibility genes; there is a familial clustering of cases, but the pattern of inheritance is not mendelian. To date, the classification of epileptic syndromes has had more of an impact on guiding clinical assessment and management than on selection of antiseizure drugs. |
Nature and Mechanisms of Seizures and Antiseizure Drugs
|
Partial Epilepsies More than a century ago, John Hughlings Jackson, the father of modern concepts of epilepsy, proposed that seizures were caused by 'occasional, sudden, excessive, rapid and local discharges of gray matter,' and that a generalized convulsion resulted when normal brain tissue was invaded by the seizure activity initiated in the abnormal focus. This insightful proposal provided a valuable framework for thinking about mechanisms of partial epilepsy. The advent of the electroencephalogram (EEG) in the 1930s permitted the recording of electrical activity from the scalp of human beings with epilepsy and demonstrated that the epilepsies are disorders of neuronal excitability. The pivotal role of synapses in mediating communication among neurons in the mammalian brain suggested that defective synaptic function might lead to a seizure. That is, a reduction of inhibitory synaptic activity or enhancement of excitatory synaptic activity might be expected to trigger a seizure; pharmacological studies of seizures supported this notion. The neurotransmitters mediating the bulk of synaptic transmission in the mammalian brain are amino acids, gamma-aminobutyric acid (GABA) and glutamate being the principal inhibitory and excitatory neurotransmitters, respectively (see Chapter 12: Neurotransmission and the Central Nervous System). Pharmacological studies disclosed that microinjection of antagonists of the GABAA receptor or of agonists of different glutamate-receptor subtypes (NMDA, AMPA, or kainic acid; see Chapter 12: Neurotransmission and the Central Nervous System) triggers seizures in experimental animals in vivo. Pharmacological agents that enhance GABA-mediated synaptic inhibition inhibit seizures in diverse models. Glutamate-receptor antagonists also inhibit seizures in diverse models, including seizures evoked by electroshock and chemical convulsants such as pentylenetetrazol. Such studies support the idea that pharmacological regulation of synaptic function can regulate the propensity for seizures, and they provide a framework for electrophysiological analyses aimed at elucidating the role of both synaptic and nonsynaptic mechanisms in expression of seizures and epilepsy. Progress in techniques of electrophysiology has fostered the progressive refinement of the level of analysis of seizure mechanisms from the EEG to populations of neurons (field potentials) to individual neurons to individual synapses and individual ion channels on individual neurons. Cellular electrophysiological studies of epilepsy over roughly two decades beginning in the mid-1960s were focused on elucidating the mechanisms underlying the depolarization shift (DS), the intracellular correlate of the 'interictal spike' (Figure 211). The interictal (or between-seizures) spike is a sharp waveform recorded in the EEG of patients with epilepsy; it is asymptomatic in that it is accompanied by no detectable change in the patient's behavior. The location of the interictal spike helps localize the brain region from which seizures originate in a given patient. The DS consists of a large depolarization of the neuronal membrane associated with a burst of action potentials. In most cortical neurons, the DS is generated by a large excitatory synaptic current that can be enhanced by activation of voltage-regulated intrinsic membrane currents (for review see Dichter and Ayala, 1987). Although the mechanisms generating the DS are increasingly understood, it remains unclear whether the interictal spike triggers a seizure, inhibits a seizure, or is an epiphenomenon with respect to seizure occurrence in an epileptic brain. While these questions remain unanswered, study of the mechanisms of DS generation set the stage for inquiry into the cellular mechanisms of a seizure.
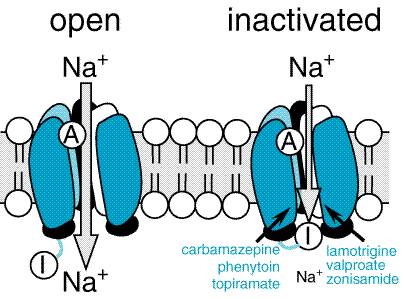
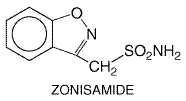
Figure 211. Relations among Cortical EEG, Extracellular, and Intracellular Recordings in a Seizure Focus Induced by Local Application of a Convulsant Agent to Mammalian Cortex. The extracellular recording was made through a high-pass filter. Note the high-frequency firing of the neuron evident in both extracellular and intracellular recording during the paroxysmal depolarization shift (PDS). (Modified from Ayala et al., 1973, with permission.) During the 1980s, a diversity of in vitro models of seizures were developed in isolated brain slice preparations, in which many synaptic connections are preserved. Electrographic events with features similar to those recorded during seizures in vivo have been produced in hippocampal slices by multiple methods, including altering ionic constituents of media bathing brain slices (for review see McNamara, 1994) such as low Ca2+, zero Mg2+, or elevated K+. The accessibility and experimental control provided by these preparations has permitted mechanistic investigations. Analyses of multiple in vitro models confirmed the importance of synaptic function in initiation of a seizure, demonstrating that subtle (e.g., 20%) reductions of inhibitory synaptic function could lead to epileptiform activity and that activation of excitatory synapses could be pivotal in initiation of a seizure. Many other important factors were identified, including the volume of the extracellular space as well as intrinsic properties of a neuron such as voltage-regulated ion channels including those gating K+, Na+, and Ca2+ ions (see Traynelis and Dingledine, 1988). Identification of these diverse synaptic and nonsynaptic factors controlling seizures in vitro provides potentially valuable pharmacological targets for regulating seizure susceptibility in vivo. An additional line of investigation has centered on understanding the mechanisms by which a normal brain is transformed into an epileptic brain. Some common forms of partial epilepsy arise months to years after injury of cortex sustained as a consequence of stroke, trauma, or other factors. An effective prophylaxis administered to patients at high risk would be highly desirable. The drugs described in this chapter provide symptomatic therapy; that is, the drugs inhibit seizures in patients with epilepsy. No effective antiepileptogenic agent has been identified. Understanding the mechanisms of epileptogenesis in cellular and molecular terms would provide a framework for development of novel therapeutic approaches. The availability of animal models provides an opportunity to investigate the underlying mechanisms. One model, termed 'kindling,' is induced by periodic administration of brief, low-intensity electrical stimulation of the amygdala or other limbic structures. Initial stimulations evoke a brief electrical seizure recorded on the EEG without behavioral change, but repeated (e.g., 10 to 20) stimulations result in progressive intensification of seizures, culminating in tonic-clonic seizures (Goddard et al., 1969). Once established, the enhanced sensitivity to electrical stimulation persists for the life of the animal. Despite the exquisite propensity to intense seizures, spontaneous seizures or a truly epileptic condition do not occur until 100 to 200 stimulations have been administered. The ease of control of kindling induction (i.e., stimulations administered at the investigator's convenience), its graded onset, and the ease of quantitating epileptogenesis (number of stimulations required to evoke tonic-clonic seizures) simplify experimental study. Pharmacological studies have demonstrated that interventions limiting activation of the NMDA subtype of glutamate receptor or the trkB-subtype of neurotrophin receptor inhibit epileptogenesis in this model. These pharmacological data provide valuable clues to the cellular and molecular mechanisms underlying epileptogenesis in this model. Two additional models are produced by administration of a convulsive chemical, kainic acid or pilocarpine, resulting in an intense limbic and tonic-clonic status epilepticus lasting hours. In both models, the fleeting episode of status epilepticus is followed weeks later by the onset of spontaneous seizures (Lemos and Cavalheiro, 1995; Longo and Mello, 1998), an intriguing parallel to the scenario of complicated febrile seizures in young children followed by the emergence of spontaneous seizures years later. In contrast to the limited or no neuronal loss characteristic of the kindling model, overt destruction of hippocampal neurons occurs in both the pilocarpine and kainate models, reflecting aspects of hippocampal sclerosis observed in human beings with severe limbic seizures. Indeed, the recent discovery that complicated febrile seizures can cause hippocampal sclerosis in young children (VanLandingham et al., 1998) establishes yet another commonality between these models and the human condition. Several questions arise with respect to these models. What transpires during the latent period between status epilepticus induced by pilocarpine or kainate and emergence of spontaneous seizures that causes the epilepsy? Might similar mechanisms be operative in kindling development and during the latent period following status epilepticus? Might an antiepileptogenic agent that was effective in one of these models be effective in the other models? Important insights into the mechanisms of action of drugs that are effective against partial seizures have emerged in the past two decades (Macdonald and Greenfield, 1997). These insights have emerged in large part from electrophysiological studies of relatively simple in vitro models, such as neurons isolated from the mammalian CNS and maintained in primary culture. The experimental control and accessibility provided by these models together with careful attention to clinically relevant concentrations of the drugs led to clarification of the mechanisms. Although it is difficult to prove unequivocally that a given drug effect observed in vitro is both necessary and sufficient to inhibit a seizure in an animal or human being in vivo, there is an excellent likelihood that the putative mechanisms identified do in fact underlie the clinically relevant antiseizure effects. Electrophysiological analyses of individual neurons during a partial seizure demonstrate that the neurons undergo depolarization and fire action potentials at high frequencies (Figure 211). This pattern of neuronal firing is characteristic of a seizure and is uncommon during physiological neuronal activity. Thus, selective inhibition of this pattern of firing would be expected to reduce seizures with minimal unwanted effects. Carbamazepine, lamotrigine, phenytoin, and valproic acid inhibit high-frequency firing at concentrations known to be effective at limiting seizures in human beings (Macdonald and Greenfield, 1997). Inhibition of the high-frequency firing is thought to be mediated by reducing the ability of Na+ channels to recover from inactivation (Figure 212). That is, depolarization-triggered opening of the Na+ channels in the axonal membrane of a neuron is required for an action potential; after opening, the channels spontaneously close, a process termed inactivation. This inactivation is thought to cause the refractory period, a short time after an action potential during which it is not possible to evoke another action potential. Upon recovery from inactivation, the Na+ channels are again poised to participate in another action potential. Because firing at a slow rate permits sufficient time for Na+ channels to recover from inactivation, inactivation has little or no effect on low-frequency firing. However, reducing the rate of recovery of Na+ channels from inactivation would limit the ability of a neuron to fire at high frequencies, an effect that likely underlies the effects of carbamazepine, lamotrigine, phenytoin, topiramate, valproic acid, and zonisamide against partial seizures.
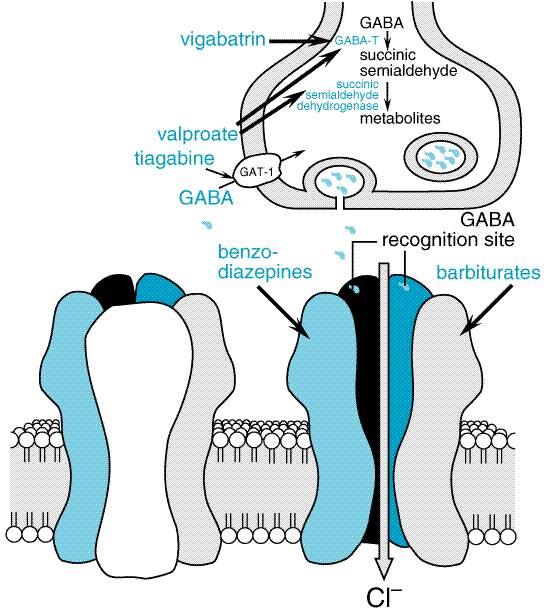
Insights
into mechanisms of seizures suggest that enhancing GABA-mediated synaptic
inhibition would reduce neuronal excitability and raise the seizure
threshold. Several drugs are thought to inhibit seizures by regulating
GABA-mediated synaptic inhibition through an action at distinct sites of the
synapse (Macdonald and Greenfield, 1997). The principal postsynaptic receptor
of synaptically released GABA is termed the GABAA receptor.
Activation of the GABAA receptor effects inhibition of the
postsynaptic cell by increasing the flow of Cl ions into the cell,
which tends to hyperpolarize the neuron. Clinically relevant concentrations
of both benzodiazepines and barbiturates can enhance GABAA
receptor-mediated inhibition through distinct actions on the GABAA
receptor (Figure 213). This mechanism probably underlies the effectiveness
of these compounds against partial and tonic-clonic seizures in human beings.
At higher concentrations, such as might be used for status epilepticus, these
drugs also can inhibit high-frequency firing of action potentials.
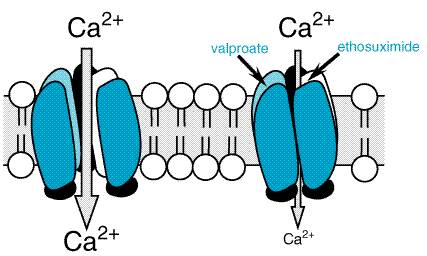
Generalized-Onset Epilepsies: Absence Seizures In contrast to partial seizures, which arise from localized regions of the cerebral cortex, generalized-onset seizures arise from the reciprocal firing of the thalamus and cerebral cortex (see Coulter, 1998, for review). Among the diverse forms of generalized seizures, absence seizures have been most intensively studied. The striking synchrony in appearance of generalized seizure discharges in widespread areas of neocortex led to the idea that a structure in the thalamus and/or brainstem (the 'centrencephalon') synchronized these seizure discharges (Penfield and Jasper, 1947). Attention on the thalamus in particular emerged from the demonstration that low-frequency stimulation of midline thalamic structures triggered EEG rhythms in the cortex similar to spike-wave discharges characteristic of absence seizures (Jasper and Droogleever-Fortuyn, 1947). Intracerebral electrode recordings from human beings subsequently demonstrated the presence of thalamic and neocortical involvement in the spike-and-wave discharge of absence seizures. Many of the structural and functional properties of thalamus and neocortex that lead to the generalized spike-and-wave discharges have been elucidated in the past decade (Coulter, 1998). The EEG hallmark of an absence seizure is generalized spike-and-wave discharges at a frequency of 3 per second. These bilaterally synchronous spike-and-wave discharges, recorded locally from electrodes in both the thalamus and the neocortex, represent oscillations between thalamus and neocortex. A comparison of EEG and intracellular recordings reveals that the EEG spikes are associated with the firing of action potentials and the following slow wave with prolonged inhibition. These reverberatory, low-frequency rhythms are made possible by a combination of factors, including reciprocal excitatory synaptic connections between neocortex and thalamus as well as intrinsic properties of neurons in the thalamus (see Coulter, 1998, for review). One intrinsic property of thalamic neurons that is pivotally involved in the generation of the 3-per-second spike and wave is a particular form of voltage-regulated Ca2+ current, the low threshold ('T') current. In contrast to its small size in most neurons, the T current in many neurons throughout the thalamus has a large amplitude. Indeed bursts of action potentials in thalamic neurons are mediated by activation of the T current. The T current plays an amplifying role in thalamic oscillations, one oscillation being the 3-per-second spike and wave of the absence seizure. Importantly, the principal mechanism by which most antiabsence-seizure drugs (ethosuximide, trimethadione, valproic acid) are thought to act is by inhibition of the T current (Figure 214; Macdonald and Kelly, 1993). Thus, inhibiting voltage-regulated ion channels is a common mechanism of action of antiseizure drugs, antipartial-seizure drugs inhibiting voltage-activated Na+ channels and antiabsence-seizure drugs inhibiting voltage-activated Ca2+ channels.
Genetic Approaches to the Epilepsies Genetic causes contribute to a wide diversity of human epilepsies. Genetic causes are solely responsible for some rare forms inherited in a mendelian patternfor example, autosomal dominant or autosomal recessive. Genetic causes also are mainly responsible for some common forms such as juvenile myoclonic epilepsy (JME) or childhood absence epilepsy (CAE), disorders likely due to inheritance of two or more susceptibility genes. Genetic determinants also may contribute some degree of risk to epilepsies caused by injury of the cerebral cortex (Berkovic, 1998). Enormous progress has been made in understanding the genetics of mammalian epilepsy in the past several years. Whereas prior to 1994, a specific gene defect had been identified in only a single mouse with a phenotype of cortical epilepsy, more than 33 single gene mutations now have been linked to an epileptic phenotype (Puranam and McNamara, 1999). This progress has been paralleled by the genetics of human epilepsy. Prior to 1990, not a single gene causing a form of human epilepsy had been identified; mutations of more than a dozen such genes now have been identified. Most of the human epilepsies for which mutant genes have been identified are symptomatic epilepsies, in which the epilepsy seems to be a manifestation of some profound neurodegenerative disease. However, most patients with epilepsy are neurologically normal. It is not clear the extent to which the mechanisms underlying the hyperexcitability in a neurologically devastating disease inform mechanisms operative in epilepsies in which the patient is otherwise normal (idiopathic epilepsies). The mutant genes have been identified in four distinct forms of idiopathic human epilepsy. Remarkably, each of the mutant genes encodes an ion channel gated by voltage or a neurotransmitter; this is of particular interest because several other episodic disorders involving other organs also are caused by mutations of genes encoding ion channels. That is, episodic disorders of the heart (cardiac arrhythmias), skeletal muscle (periodic paralyses), cerebellum (episodic ataxia), vasculature (familial hemiplegic migraine), and other organs all have been linked to mutant genes encoding a component of a voltage-gated ion channel (Ptacek, 1997). The four idiopathic human epilepsies for which the mutant genes have
been identified are the following. Generalized epilepsy with febrile seizures
(GEFS+) is caused by a point mutation in the |
Antiseizure Drugs: General Considerations
|
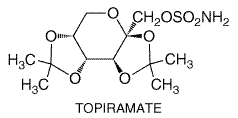
History Phenobarbital was the first synthetic organic agent recognized as having antiseizure activity (Hauptmann, 1912); its sedative properties led investigators to test and demonstrate its effectiveness for suppressing seizures. In a landmark discovery, Merritt and Putnam (1938a) developed the electroshock seizure test in experimental animals to screen chemical agents for antiseizure effectiveness; in the course of screening a variety of drugs, they discovered that phenytoin suppressed seizures in the absence of sedative effects. The electroshock seizure test is extremely valuable, because drugs that are effective against tonic hindlimb extension induced by electroshock generally have proven to be effective against partial and tonic-clonic seizures in human beings. Another screening test, seizures induced by the chemoconvulsant pentylenetetrazol, is useful in identifying drugs that are effective against absence seizures in human beings. These screening tests remain useful even now. The chemical structures of most of the drugs introduced before 1965 were closely related to phenobarbital. These include the hydantoins, the oxazolidinediones, and the succinimides. The agents introduced after 1965 exhibit a diversity of chemical structures. These include benzodiazepines (clonazepam and clorazepate), an iminostilbene (carbamazepine), a branched-chain carboxylic acid (valproic acid), a phenyltriazine (lamotrigine), a cyclic analog of GABA (gabapentin), a sulfamate-substituted monosaccharide (topiramate), a nipecotic acid derivative (tiagabine), and a pyrrolidine derivative (levetiracetam). Therapeutic Aspects The ideal antiseizure drug would suppress all seizures without causing any unwanted effects. Unfortunately, the drugs used currently not only fail to control seizure activity in some patients, but they frequently cause unwanted effects that range in severity from minimal impairment of the CNS to death from aplastic anemia or hepatic failure. The physician who treats patients with epilepsy is thus faced with the task of selecting the appropriate drug or combination of drugs that best controls seizures in an individual patient at an acceptable level of untoward effects. It is generally held that complete control of seizures can be achieved in up to 50% of patients and that another 25% can be improved significantly. The degree of success varies as a function of seizure type, cause, and other factors. To minimize toxicity, treatment with a single drug should be sought. If seizures are not controlled at adequate plasma concentrations of the initial agent, substitution of a second drug is preferred to the concurrent administration of another agent. However, multiple-drug therapy may be required, especially when two or more types of seizure occur in the same patient. Measurement of drug concentrations in plasma facilitates optimizing antiseizure medication, especially when therapy is initiated, after dosage adjustments, in the event of therapeutic failure, when toxic effects appear, or when multiple-drug therapy is instituted. However, clinical effects of some drugs do not correlate well with their concentrations in plasma, and recommended concentrations are only guidelines for therapy. The ultimate therapeutic regimen must be determined by clinical assessment of effect and toxicity. The general principles of the drug therapy of the epilepsies are summarized below, following discussion of the individual agents. Details of diagnosis and therapy can be found in the monographs and reviews listed at the end of the chapter. |
Hydantoins
|
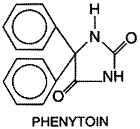
Phenytoin Phenytoin (diphenylhydantoin; DILANTIN DIPHENYLAN) is effective against all types of partial and tonic-clonic seizures but not absence seizures. Properties of other hydantoins (mephenytoin, ethotoin) are described in earlier editions of this book. History Phenytoin was first synthesized in 1908 by Biltz, but its anticonvulsant activity was not discovered until 1938 (Merritt and Putnam, 1938a,b). In contrast to the earlier accidental discovery of the antiseizure properties of bromide and phenobarbital, phenytoin was the product of a search among nonsedative structural relatives of phenobarbital for agents capable of suppressing electroshock convulsions in laboratory animals. It was introduced for the treatment of epilepsy in the same year. The discovery of phenytoin was a signal advance. Since this agent is not a sedative in ordinary doses, it established that antiseizure drugs need not induce drowsiness and encouraged the search for drugs with selective antiseizure action. Structure-Activity Relationship Phenytoin has the following structural formula:
A 5-phenyl or other aromatic substituent appears essential for activity against generalized tonic-clonic seizures. Alkyl substituents in position 5 contribute to sedation, a property absent in phenytoin. The position 5 carbon permits asymmetry, but there appears to be little difference in activity between isomers. Pharmacological Effects Central Nervous System Phenytoin exerts antiseizure activity without causing general depression of the CNS. In toxic doses it may produce excitatory signs and at lethal levels a type of decerebrate rigidity. The most significant effect of phenytoin is its ability to modify the pattern of maximal electroshock seizures. The characteristic tonic phase can be abolished completely, but the residual clonic seizure may be exaggerated and prolonged. This seizure-modifying action is observed with many other antiseizure drugs that are effective against generalized tonic-clonic seizures. By contrast, phenytoin does not inhibit clonic seizures evoked by pentylenetetrazol. Mechanism of Action Phenytoin limits the repetitive firing of action potentials evoked by a sustained depolarization of mouse spinal cord neurons maintained in vitro (McLean and Macdonald, 1983). This effect is mediated by a slowing of the rate of recovery of voltage-activated Na+ channels from inactivation, an action that is both voltage- (greater effect if membrane is depolarized) and use-dependent. These effects of phenytoin are evident at concentrations in the range of therapeutic drug levels in cerebrospinal fluid (CSF) in human beings, concentrations that correlate with the free (or unbound) concentration of phenytoin in the serum. At these concentrations, the effects on Na+ channels are selective, in that no changes of spontaneous activity or responses to iontophoretically-applied GABA or glutamate are detected. At concentrations 5- to 10-fold higher, multiple effects of phenytoin are evident, including reduction of spontaneous activity, enhancement of responses to GABA, and others; these effects may underlie some of the unwanted toxicity associated with high levels of phenytoin. Pharmacokinetic Properties The pharmacokinetic characteristics of phenytoin are influenced markedly by its binding to serum proteins, by the nonlinearity of its elimination kinetics, and by its metabolism by the cytochrome P450 enzyme system. Phenytoin is extensively bound (about 90%) to serum proteins, mainly albumin. Small variations in the percentage of phenytoin that is bound dramatically affect the absolute amount of free (active) drug; increased proportions of free drug are evident in the neonate, in patients with hypoalbuminemia, and in uremic patients. Some agents, such as valproate, can compete with phenytoin for binding sites on plasma proteins; when combined with valproate-mediated inhibition of phenytoin metabolism, marked increases in free phenytoin can result. Measurement of free rather than total phenytoin permits direct assessment of this potential problem in patient management. Phenytoin is one of the few drugs for which the rate of elimination
varies as a function of its concentration (i.e., the rate is
nonlinear). The plasma half-life of phenytoin ranges between 6 and 24 hours
at plasma concentrations below 10 The majority (95%) of phenytoin is metabolized principally in the hepatic endoplasmic reticulum and mainly by the cytochrome P450 isoform CYP2C9/10 and to a lesser extent CYP2C19 (Table 212). The principal metabolite, a parahydroxyphenyl derivative, is inactive. Because its metabolism is saturable, other drugs that are metabolized by these enzymes can inhibit phenytoin's metabolism and produce a rise in phenytoin concentration. Conversely, the degradation rate of other drugs that are substrates for these enzymes can be inhibited by phenytoin; one such drug is warfarin, and addition of phenytoin to a patient receiving warfarin can lead to hypoprothrombinemia. An alternative mechanism of drug interactions arises from phenytoin's ability to induce diverse cytochrome P450 enzymes; coadministration of phenytoin and medications metabolized by these enzymes can lead to an increased degradation of such medications. Of particular note in this regard are oral contraceptives, which are metabolized by the CYP3A4; treatment with phenytoin could enhance the metabolism of oral contraceptives and lead to unplanned pregnancy. The potential teratogenic effects of phenytoin underscore the importance of attention to this interaction. Carbamazepine, oxcarbazepine, phenobarbital, and primidone also can induce CYP3A4 and likewise might increase degradation of oral contraceptives. The low aqueous solubility of phenytoin resulted in diverse problems for intravenous use and led to production of fosphenytoin, a water-soluble prodrug. Fosphenytoin (CEREBYX) is converted into phenytoin by phosphatases in liver and red blood cells with a half-life of 8 to 15 minutes. Fosphenytoin is useful for adults with partial or generalized seizures when parenteral administration is indicated. Toxicity The toxic effects of phenytoin depend upon the route of administration, the duration of exposure, and the dosage. When fosphenytoin, the water-soluble prodrug, is administered intravenously at an excessive rate in the emergency treatment of status epilepticus, the most notable toxic signs are cardiac arrhythmias, with or without hypotension, and/or CNS depression. Although cardiac toxicity occurs more frequently in older patients and in those with known cardiac disease, it also can develop in young, healthy patients. These complications can be minimized by administering fosphenytoin at a rate of less than 150 mg of phenytoin sodium equivalents per minute. Acute oral overdosage results primarily in signs referable to the cerebellum and vestibular system; high doses have been associated with marked cerebellar atrophy. Toxic effects associated with chronic medication also are primarily dose-related cerebellar-vestibular effects but include other CNS effects, behavioral changes, increased frequency of seizures, gastrointestinal symptoms, gingival hyperplasia, osteomalacia, and megaloblastic anemia. Hirsutism is an annoying untoward effect in young females. Usually, these phenomena can be made bearable by proper adjustment of dosage. Serious adverse effects, including those on the skin, bone marrow, and liver, probably are manifestations of drug allergy. Although rare, they necessitate withdrawal of the drug. Moderate elevation of the concentrations in plasma of enzymes that are used to assess hepatic function sometimes are observed; since these changes are transient and may result in part from induced synthesis of the enzymes, they do not necessitate withdrawal of the drug. Electrophysiological evidence of peripheral neuropathy can occur in up to 30% of patients receiving phenytoin, but this phenomenon usually is not clinically significant. Gingival hyperplasia occurs in about 20% of all patients during chronic therapy and is probably the most common manifestation of phenytoin toxicity in children and young adolescents. It may be more frequent in those individuals who also develop coarsened facial features. The overgrowth of tissue appears to involve altered collagen metabolism. Toothless portions of the gums are not affected. The condition does not necessarily require withdrawal of medication, and it can be minimized by good oral hygiene. A variety of endocrine effects have been reported. Inhibition of release of antidiuretic hormone (ADH) has been observed in patients with inappropriate ADH secretion. Hyperglycemia and glycosuria appear to be due to inhibition of insulin secretion. Osteomalacia, with hypocalcemia and elevated alkaline phosphatase activity, has been attributed to both altered metabolism of vitamin D and inhibition of intestinal absorption of Ca2+. Phenytoin also increases the metabolism of vitamin K and reduces the concentration of vitamin Kdependent proteins that are important for normal Ca2+ metabolism in bone. This may explain why the osteomalacia is not always ameliorated by the administration of vitamin D. Hypersensitivity reactions include morbilliform rash in 2% to 5% of patients and occasionally more serious skin reactions, including Stevens-Johnson syndrome. Systemic lupus erythematosus and potentially fatal hepatic necrosis have been reported rarely. Hematological reactions include neutropenia and leukopenia. A few instances of red-cell aplasia, agranulocytosis, and mild thrombocytopenia also have been reported. Lymphadenopathy, resembling Hodgkin's disease and malignant lymphoma, is associated with reduced immunoglobulin A (IgA) production. Hypoprothrombinemia and hemorrhage have occurred in the newborns of mothers who received phenytoin during pregnancy; vitamin K is effective treatment or prophylaxis. Plasma Drug Concentrations A good correlation usually is observed between the total concentration
of phenytoin in plasma and the clinical effect. Thus, control of seizures
generally is obtained with concentrations above 10 Drug Interactions Concurrent administration of any drug metabolized by the 2C9/10 isoform of cytochrome P450 can increase the plasma concentration of phenytoin by decreasing its rate of metabolism. Carbamazepine, which may enhance the metabolism of phenytoin, causes a well-documented decrease in phenytoin concentration. Conversely, phenytoin reduces the concentration of carbamazepine. Interaction between phenytoin and phenobarbital is variable. Therapeutic Uses Epilepsy Phenytoin is one of the more widely used antiseizure agents, and it is effective against partial and tonic-clonic but not absence seizures. The use of phenytoin and other agents in the therapy of epilepsies is discussed further at the end of this chapter. Various preparations of phenytoin differ significantly in both bioavailability and rate of absorption, and patients should thus be treated with the drug product of a single manufacturer. Other Uses Some cases of trigeminal and related neuralgias appear to respond to phenytoin, but carbamazepine may be preferable. The use of phenytoin in the treatment of cardiac arrhythmias is discussed in Chapter 35: Antiarrhythmic Drugs. |
Antiseizure Barbiturates
|
The pharmacology of the barbiturates as a class is considered in Chapter 17: Hypnotics and Sedatives; discussion in this chapter is limited to the two barbiturates used for therapy of the epilepsies. Although still marketed, a third barbiturate (metharbital) has virtually disappeared from therapeutic use. Phenobarbital Phenobarbital LUMINAL, others) was the first effective organic antiseizure agent (Hauptmann, 1912). It has relatively low toxicity, is inexpensive, and is still one of the more effective and widely used drugs for this purpose. Structure-Activity Relationship The structural formula of phenobarbital (5-phenyl-5-ethylbarbituric acid) is shown in Chapter 17: Hypnotics and Sedatives. The structure-activity relationship of the barbiturates has been studied extensively. Maximal antiseizure activity is obtained when one substituent at position 5 is a phenyl group. The 5,5-diphenyl derivative has less antiseizure potency than does phenobarbital, but it is virtually devoid of hypnotic activity. By contrast, 5,5-dibenzyl barbituric acid causes convulsions. Antiseizure Properties Most barbiturates have antiseizure properties. However, the capacity of some of these agents, such as phenobarbital, to exert maximal antiseizure action at doses below those required for hypnosis determines their clinical utility as antiseizure agents. Phenobarbital is active in most antiseizure tests in animals but is relatively nonselective. It inhibits tonic hindlimb extension in the maximal electroshock model, clonic seizures evoked by pentylenetetrazol, and kindled seizures. Mechanism of Action The mechanism by which phenobarbital inhibits seizures likely involves potentiation of synaptic inhibition through an action on the GABAA receptor. Intracellular recordings of mouse cortical or spinal cord neurons demonstrated that phenobarbital enhances responses to iontophoretically applied GABA (Macdonald and Barker, 1979). These effects have been observed at therapeutically relevant concentrations of phenobarbital. Analyses of single channels in outside-out patches isolated from mouse spinal cord neurons demonstrated that phenobarbital increased the GABA receptormediated current by increasing the duration of bursts of GABA receptormediated currents without changing the frequency of bursts (Twyman et al., 1989). At levels exceeding therapeutic concentrations, phenobarbital also limits sustained repetitive firing; this may underlie some of the antiseizure effects of higher concentrations of phenobarbital achieved during therapy of status epilepticus. The mechanisms underlying the antiseizure as opposed to the sedative effects of the barbiturates have been enigmatic. That is, pentobarbital inhibits seizures, but at doses that produce marked sedation; by contrast, phenobarbital inhibits seizures at doses that cause minimal sedative effects. Both pentobarbital and phenobarbital enhance GABAA receptormediated currents. Distinctive effects of pentobarbital and phenobarbital on GABA responses and voltage-activated Ca2+ channels may explain this enigma (ffrench-Mullen et al., 1993). The maximal effect of phenobarbital in enhancing GABA responses is only 40% of that of the active isomer of pentobarbital. Moreover, pentobarbital inhibits voltage-activated Ca2+ channels with greater potency than does phenobarbital (ffrench-Mullen et al., 1993); one consequence of inhibition of these Ca2+ channels could be blockade of Ca2+ entry into presynaptic nerve terminals and inhibition of release of neurotransmitters such as glutamate, resulting in net reduction of excitatory synaptic transmission. Thus the powerful sedative actions of pentobarbital could be due to greater maximal enhancement of GABA responses in conjunction with strong inhibition of Ca2+ current. Pharmacokinetic Properties Oral absorption of phenobarbital is complete but somewhat slow; peak concentrations in plasma occur several hours after a single dose. It is 40% to 60% bound to plasma proteins and bound to a similar extent in tissues, including brain. Up to 25% of a dose is eliminated by pH-dependent renal excretion of the unchanged drug; the remainder is inactivated by hepatic microsomal enzymes. The principal cytochrome P450 responsible is CYP2C9, with minor metabolism by CYP2C19 and 2E1. Phenobarbital induces uridine diphosphate glucuronosyl transferase (UGT) enzymes as well as CYP2C and 3A subfamilies of cytochrome P450. Drugs metabolized by these enzymes can be more rapidly degraded when coadministered with phenobarbital; importantly, oral contraceptives are metabolized by CYP3A4. Toxicity Sedation, the most frequent undesired effect of phenobarbital, is apparent to some extent in all patients upon initiation of therapy, but tolerance develops during chronic medication. Nystagmus and ataxia occur at excessive dosage. Phenobarbital sometimes produces irritability and hyperactivity in children, and agitation and confusion in the elderly. Scarlatiniform or morbilliform rash, possibly with other manifestations of drug allergy, occurs in 1% to 2% of patients. Exfoliative dermatitis is rare. Hypoprothrombinemia with hemorrhage has been observed in the newborn of mothers who have received phenobarbital during pregnancy; vitamin K is effective for treatment or prophylaxis. Megaloblastic anemia that responds to folate and osteomalacia that responds to high doses of vitamin D occur during chronic phenobarbital therapy of epilepsy, as they do during phenytoin medication. Other adverse effects of phenobarbital are discussed in Chapter 17: Hypnotics and Sedatives. Plasma Drug Concentrations During long-term therapy in adults, the plasma concentration of phenobarbital
averages 10 The relationship between plasma concentration of phenobarbital and
adverse effects varies with the development of tolerance. Sedation,
nystagmus, and ataxia usually are absent at concentrations below 30 Since significant behavioral toxicity may be present despite the
absence of overt signs of toxicity, the tendency to maintain patients,
particularly children, on excessively high doses of phenobarbital should be
resisted. The plasma phenobarbital concentration should be increased above 30
to 40 Drug Interactions Interactions between phenobarbital and other drugs usually involve induction of the hepatic microsomal enzyme system by phenobarbital (see Chapters 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination and 17: Hypnotics and Sedatives). The variable interaction with phenytoin has been discussed above. Concentrations of phenobarbital in plasma may be elevated by as much as 40% during concurrent administration of valproic acid (see below). Therapeutic Uses Phenobarbital is an effective agent for generalized tonic-clonic and partial seizures. Its efficacy, low toxicity, and low cost make it an important agent for these types of epilepsy. However, its sedative effects and its tendency to disturb behavior in children have reduced its use as a primary agent. Mephobarbital MEBARAL) is N-methylphenobarbital. It is N-demethylated in the hepatic endoplasmic reticulum, and most of its activity during long-term therapy can be attributed to the accumulation of phenobarbital. Consequently, the pharmacological properties, toxicity, and clinical uses of mephobarbital are the same as those for phenobarbital. |
Deoxybarbiturates
|
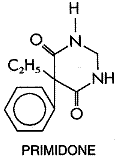
Primidone Primidone MYSOLINE) is effective against partial and tonic-clonic seizures. Chemistry Primidone may be viewed as a congener of phenobarbital in which the carbonyl oxygen of the urea moiety is replaced by two hydrogen atoms:
Antiseizure Properties Primidone resembles phenobarbital in many laboratory antiseizure effects, but it is much less potent than phenobarbital in antagonizing seizures induced by pentylenetetrazol. The antiseizure effects of primidone are attributed to both the drug and its active metabolites, principally phenobarbital. Pharmacokinetic Properties Primidone is rapidly and almost completely absorbed after oral administration, although individual variability can be great. Peak concentrations in plasma usually are observed approximately 3 hours after ingestion. The plasma half-life of primidone is variable; mean values ranging from 5 to 15 hours have been reported. Primidone is converted to two active metabolites, phenobarbital and phenylethylmalonamide (PEMA). Primidone and PEMA are bound to plasma proteins to only a small extent, whereas about half of phenobarbital is so bound. The half-life of PEMA in plasma is 16 hours; both it and phenobarbital accumulate during long-term therapy. The appearance of phenobarbital in plasma may be delayed several days upon initiation of therapy with primidone. Approximately 40% of the drug is excreted unchanged in the urine; unconjugated PEMA and, to a lesser extent, phenobarbital and its metabolites constitute the remainder. Toxicity The more common complaints are sedation, vertigo, dizziness, nausea, vomiting, ataxia, diplopia, and nystagmus. Patients also may experience an acute feeling of intoxication immediately following administration of primidone. This occurs before there is any significant metabolism of the drug. The relationship of adverse effects to dosage is complex, since they result from both the parent drug and its two active metabolites and since tolerance develops during long-term therapy. Side effects are occasionally quite severe when therapy is initiated. Serious adverse effects are relatively uncommon, but maculopapular and morbilliform rash, leukopenia, thrombocytopenia, systemic lupus erythematosus, and lymphadenopathy have been reported. Acute psychotic reactions also have occurred. Hemorrhagic disease in the neonate, megaloblastic anemia, and osteomalacia similar to those discussed previously in connection with phenytoin and phenobarbital also have been described. Plasma Drug Concentrations The relationship between the dose of primidone and the concentration
of the drug and its active metabolites in plasma shows marked individual
variability. During long-term therapy, the plasma concentrations of primidone
and phenobarbital average 1 Drug Interactions Phenytoin has been reported to increase the conversion of primidone to phenobarbital. Other drug interactions to be anticipated are those for phenobarbital. Therapeutic Uses Primidone is useful against generalized tonic-clonic and both simple and complex partial seizures. Its use in combination with phenobarbital is illogical. Primidone is ineffective against absence seizures but is sometimes useful against myoclonic seizures in young children. |
Iminostilbenes
|
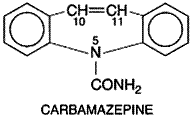
Carbamazepine Carbamazepine TEGRETOL, CARBATROL, others) was initially approved in the Chemistry Carbamazepine is related chemically to the tricyclic antidepressants. It is a derivative of iminostilbene with a carbamyl group at the 5 position; this moiety is essential for potent antiseizure activity. The structural formula of carbamazepine is as follows:
Pharmacological Effects Although the effects of carbamazepine in animals and human beings resemble those of phenytoin in many ways, the two drugs differ in a number of potentially important ways. Carbamazepine has been found to produce therapeutic responses in manic-depressive patients, including some in whom lithium carbonate is not effective. Further, carbamazepine has antidiuretic effects that are sometimes associated with reduced concentrations of antidiuretic hormone (ADH) in plasma. The mechanisms responsible for these effects of carbamazepine are not clearly understood. Mechanism of Action Like phenytoin, carbamazepine limits the repetitive firing of action potentials evoked by a sustained depolarization of mouse spinal cord or cortical neurons maintained in vitro (McLean and Macdonald, 1986b). This appears to be mediated by a slowing of the rate of recovery of voltage-activated Na+ channels from inactivation. These effects of carbamazepine are evident at concentrations in the range of therapeutic drug levels in CSF in human beings. The effects of carbamazepine are selective at these concentrations, in that there are no effects on spontaneous activity or on responses to iontophoretically applied GABA or glutamate. The carbamazepine metabolite, 10,11-epoxycarbamazepine, also limits sustained repetitive firing at therapeutically relevant concentrations, suggesting that this metabolite may contribute to the antiseizure efficacy of carbamazepine. Pharmacokinetic Properties The pharmacokinetic characteristics of carbamazepine are complex. They are influenced by its limited aqueous solubility and by the ability of many antiseizure drugs, including carbamazepine itself, to increase their conversion to active metabolites by hepatic oxidative enzymes. Carbamazepine is absorbed slowly and erratically after oral administration. Peak concentrations in plasma usually are observed 4 to 8 hours after oral ingestion, but may be delayed by as much as 24 hours, especially following the administration of a large dose. The drug distributes rapidly into all tissues. Binding to plasma proteins occurs to the extent of about 75%, and concentrations in the CSF appear to correspond to the concentration of free drug in plasma. The predominant pathway of metabolism in human beings involves conversion to the 10,11-epoxide. This metabolite is as active as the parent compound in various animals, and its concentrations in plasma and brain may reach 50% of those of carbamazepine, especially during the concurrent administration of phenytoin or phenobarbital. The 10,11-epoxide is metabolized further to inactive compounds, which are excreted in the urine principally as glucuronides. Carbamazepine also is inactivated by conjugation and hydroxylation. The hepatic cytochrome P450 isoform primarily responsible for biotransformation of carbamazepine is CYP3A4. Carbamazepine induces CYP2C and CYP3A and also UDP-glucuronosyltransferase, thus enhancing the metabolism of drugs degraded by these enzymes. Of particular importance in this regard are oral contraceptives, which are metabolized by CYP3A4. Toxicity Acute intoxication with carbamazepine can result in stupor or coma, hyperirritability, convulsions, and respiratory depression. During long-term therapy, the more frequent untoward effects of the drug include drowsiness, vertigo, ataxia, diplopia, and blurred vision. The frequency of seizures may increase, especially with overdosage. Other adverse effects include nausea, vomiting, serious hematological toxicity (aplastic anemia, agranulocytosis), and hypersensitivity reactions (dermatitis, eosinophilia, lymphadenopathy, splenomegaly). A late complication of therapy with carbamazepine is retention of water, with decreased osmolality and concentration of Na+ in plasma, especially in elderly patients with cardiac disease. Some tolerance develops to the neurotoxic effects of carbamazepine, and they can be minimized by gradual increase in dosage or adjustment of maintenance dosage. Various hepatic or pancreatic abnormalities have been reported during therapy with carbamazepine, most commonly a transient elevation of hepatic enzymes in plasma in 5% to 10% of patients. A transient, mild leukopenia occurs in about 10% of patients during initiation of therapy and usually resolves within the first 4 months of continued treatment; transient thrombocytopenia also has been noted. In about 2% of patients, a persistent leukopenia may develop that requires withdrawal of the drug. The initial concern that aplastic anemia might be a frequent complication of long-term therapy with carbamazepine has not materialized. In the majority of cases, the administration of multiple drugs or the presence of another underlying disease has made it difficult to establish a causal relationship. In any event, the prevalence of aplastic anemia appears to be about 1 in 200,000 patients who are treated with the drug. It is not clear whether or not monitoring of hematological function can avert the development of irreversible aplastic anemia. Although carbamazepine is carcinogenic in rats, it is not known to be carcinogenic in human beings. The induction of fetal malformations during the treatment of pregnant women is discussed below. Plasma Drug Concentrations There is no simple relationship between the dose of carbamazepine and
concentrations of the drug in plasma. Therapeutic concentrations are reported
to be 6 to 12 Drug Interactions Phenobarbital, phenytoin, and valproate may increase the metabolism of carbamazepine by inducing CYP3A4; carbamazepine may enhance the biotransformation of phenytoin as well as the conversion of primidone to phenobarbital. Administration of carbamazepine may lower concentrations of valproate, lamotrigine, tiagabine, and topiramate given concurrently. Carbamazepine reduces both the plasma concentration and therapeutic effect of haloperidol. The metabolism of carbamazepine may be inhibited by propoxyphene, erythromycin, cimetidine, fluoxetine, and isoniazid. Therapeutic Uses Carbamazepine is useful in patients with generalized tonic-clonic and both simple and complex partial seizures. When it is used, renal and hepatic function and hematological parameters should be monitored. The therapeutic use of carbamazepine is discussed further at the end of this chapter. Carbamazepine was introduced by Blom in the early 1960s and is now the primary agent for treatment of trigeminal and glossopharyngeal neuralgias. It is also effective for lightning tabetic pain. Most patients with neuralgia are benefited initially, but only 70% obtain continuing relief. Adverse effects have required discontinuation of medication in 5% to 20% of patients. The therapeutic range of plasma concentrations for antiseizure therapy serves as a guideline for its use in neuralgia. Carbamazepine also has found use in the treatment of bipolar affective disorders, a use that is discussed further in Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania. Oxcarbazepine Oxcarbazepine TRILEPTAL) (10,11-dihydro-10-oxocarbamazepine) is a keto analog of carbamazepine. In human beings, oxcarbazepine functions as a prodrug, in that it is almost immediately converted to its main active metabolite, a 10-monohydroxy derivative which is inactivated by glucuronide conjugation and eliminated by renal excretion. Its mechanism of action is similar to that of carbamazepine. Oxcarbazepine is a less potent enzyme inducer than is carbamazepine, and substitution of oxcarbazepine for carbamazepine is associated with increased levels of phenytoin and valproic acid, presumably because of reduced induction of hepatic enzymes. There is no induction by oxcarbazepine of hepatic enzymes involved in its degradation. Although oxcarbazepine does not appear to reduce the anticoagulant effect of warfarin, it does induce CYP3A and thus reduces plasma levels of steroid oral contraceptives. It has been approved for monotherapy or adjunct therapy for partial seizures in adults and as adjunctive therapy for partial seizures in children ages 4 to 16. |
Succinimides
|
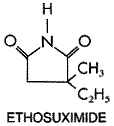
Ethosuximide The succinimides evolved from a systematic search for effective agents less toxic than the oxazolidinediones for the treatment of absence seizures. Ethosuximide (ZARONTIN) is a primary agent for this type of epilepsy. Structure-Activity Relationship Ethosuximide has the following structural formula:
The structure-activity relationship of the succinimides is in accord with that for other antiseizure classes. Methsuximide (CELONTIN) and phensuximide (MILONTIN) have phenyl substituents and are more active against maximal electroshock seizures. Neither of these is now in common use. Discussion of their properties can be found in older editions of this book. Ethosuximide, with alkyl substituents, is the most active of the succinimides against seizures induced by pentylenetetrazol and is the most selective for absence seizures. Pharmacological Effects The most prominent characteristic of ethosuximide at nontoxic doses is protection against clonic motor seizures induced by pentylenetetrazol. By contrast, at nontoxic doses, ethosuximide does not inhibit tonic hindlimb extension of electroshock seizures or kindled seizures. This profile correlates with efficacy against absence seizures in human beings. Mechanism of Action Ethosuximide reduces low threshold Ca2+ currents (T currents) in thalamic neurons (Coulter et al., 1989). The thalamus plays an important role in generation of 3-Hz spike-wave rhythms typical of absence seizures (Coulter, 1998). Neurons in the thalamus exhibit a large amplitude T-current spike that underlies bursts of action potentials and likely plays an important role in thalamic oscillatory activity such as 3-Hz spike-and-wave activity. At clinically relevant concentrations, ethosuximide inhibits the T current, as evident in voltage-clamp recordings of acutely isolated, ventrobasal thalamic neurons from rats and guinea pigs. Ethosuximide reduces this current without modifying the voltage dependence of steady-state inactivation or the time course of recovery from inactivation. By contrast, succinimide derivatives with convulsant properties do not inhibit this current. Ethosuximide does not inhibit sustained repetitive firing or enhance GABA responses at clinically relevant concentrations. Current data are consistent with the idea that inhibition of T currents is the mechanism by which ethosuximide inhibits absence seizures. Pharmacokinetic Properties Absorption of ethosuximide appears to be complete, and peak concentrations occur in plasma within about 3 hours after a single oral dose. Ethosuximide is not significantly bound to plasma proteins; during long-term therapy, the concentration in the CSF is similar to that in plasma. The apparent volume of distribution averages 0.7 liter/kg. In human beings, 25% of the drug is excreted unchanged in the urine. The remainder is metabolized by hepatic microsomal enzymes, but whether or not cytochrome P450 enzymes are responsible is unknown. The major metabolite, the hydroxyethyl derivative, accounts for about 40% of administered drug, is inactive, and is excreted as such and as the glucuronide in the urine. The plasma half-life of ethosuximide averages between 40 and 50 hours in adults and approximately 30 hours in children. Toxicity The most common dose-related side effects are gastrointestinal complaints (nausea, vomiting, and anorexia) and CNS effects (drowsiness, lethargy, euphoria, dizziness, headache, and hiccough). Some tolerance to these effects develops. Parkinson-like symptoms and photophobia also have been reported. Restlessness, agitation, anxiety, aggressiveness, inability to concentrate, and other behavioral effects have occurred primarily in patients with a prior history of psychiatric disturbance. Urticaria and other skin reactions, including Stevens-Johnson syndromeas well as systemic lupus erythematosus, eosinophilia, leukopenia, thrombocytopenia, pancytopenia, and aplastic anemiaalso have been attributed to the drug. The leukopenia may be transient despite continuation of the drug, but several deaths have resulted from bone-marrow depression. Renal or hepatic toxicity has not been reported. Plasma Drug Concentrations During long-term therapy, the plasma concentration of ethosuximide
averages about 2 Therapeutic Uses Ethosuximide is effective against absence seizures but not tonic-clonic seizures and has a lower risk of adverse effects than does trimethadione, a drug formerly used to treat absence seizures (its properties are discussed in earlier editions of this book). It is an important therapeutic agent for this type of epilepsy. An initial daily dose of 250 mg in children (3 to 6 years old) and 500 mg in older children and adults is increased by 250-mg increments at weekly intervals until seizures are adequately controlled or toxicity intervenes. Divided dosage is required occasionally to prevent nausea or drowsiness associated with single daily dosage. The usual maintenance dose is 20 mg/kg per day. Increased caution is required if the daily dose exceeds 1500 mg in adults or 750 to 1000 mg in children. The use of ethosuximide and the other antiseizure agents is discussed further at the end of the chapter. |
Valproic Acid
|
Valproic acid DEPAKENE, others) was approved for use in
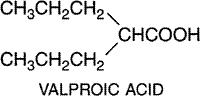
the Chemistry Valproic acid (n-dipropylacetic acid) is a simple branched-chain carboxylic acid; its structural formula is as follows:
Certain other branched-chain carboxylic acids have potencies similar to that of valproic acid in antagonizing pentylenetetrazol-induced convulsions. However, increasing the number of carbon atoms to nine introduces marked sedative properties. Straight-chain acids have little or no activity. The primary amide of valproic acid is about twice as potent as the parent compound. Pharmacological Effects Valproic acid is strikingly different from phenytoin or ethosuximide in that it is effective in inhibiting seizures in a variety of models. Like phenytoin and carbamazepine, valproate inhibits tonic hindlimb extension in maximal electroshock seizures and kindled seizures at doses without toxicity. Like ethosuximide, valproic acid inhibits clonic motor seizures induced by pentylenetetrazol at subtoxic doses. Its efficacy in diverse models parallels its efficacy against absence as well as partial and generalized tonic-clonic seizures in human beings. Mechanism of Action Valproic acid produces effects on isolated neurons similar to those of both phenytoin and ethosuximide. At therapeutically relevant concentrations, valproate inhibits sustained repetitive firing induced by depolarization of mouse cortical or spinal cord neurons (McLean and Macdonald, 1986a). The action is similar to that of both phenytoin and carbamazepine and appears to be mediated by a prolonged recovery of voltage-activated Na+ channels from inactivation. Valproic acid does not modify neuronal responses to iontophoretically applied GABA. In neurons isolated from a distinct region, the nodose ganglion, valproate also produces small reductions of the low-threshold (T) Ca2+ current (Kelly et al., 1990) at clinically relevant but slightly higher concentrations than limit sustained repetitive firing; this effect on T currents is similar to that of ethosuximide in thalamic neurons (Coulter et al., 1989). Together, these actions of limiting sustained repetitive firing and reducing T currents may contribute to the effectiveness of valproic acid against partial and tonic-clonic seizures and absence seizures, respectively. Another potential mechanism that may contribute to valproate's antiseizure actions involves metabolism of GABA. Although valproate has no effect on responses to GABA, it does increase the amount of GABA that can be recovered from the brain after the drug is administered to animals. In vitro, valproate can stimulate the activity of the GABA synthetic enzyme, glutamic acid decarboxylase (Phillips and Fowler, 1982), and inhibit GABA degradative enzymes, GABA transaminase and succinic semialdehyde dehydrogenase (Chapman et al., 1982). Thus far it has been difficult to relate the increased GABA levels to the antiseizure activity of valproate. Pharmacokinetic Properties Valproic acid is absorbed rapidly and completely after oral administration. Peak concentration in plasma is observed in 1 to 4 hours, although this can be delayed for several hours if the drug is administered in enteric-coated tablets or is ingested with meals. The apparent volume of distribution for valproate is about 0.2 liter/kg. Its extent of binding to plasma proteins is usually about 90%, but the fraction bound is reduced as the total concentration of valproate is increased through the therapeutic range. Although concentrations of valproate in CSF suggest equilibration with free drug in the blood, there is evidence for carrier-mediated transport of valproate both into and out of the CSF. The vast majority of valproate (95%) undergoes hepatic metabolism,
with less than 5% excreted unchanged. Its hepatic metabolism occurs mainly by
UGT enzymes and Toxicity The most common side effects are transient gastrointestinal symptoms, including anorexia, nausea, and vomiting in about 16% of patients. Effects on the CNS include sedation, ataxia, and tremor; these symptoms occur infrequently and usually respond to a decrease in dosage. Rash, alopecia, and stimulation of appetite have been observed occasionally. Valproic acid has several effects on hepatic function. Elevation of hepatic enzymes in plasma is observed in up to 40% of patients and often occurs asymptomatically during the first several months of therapy. A rare complication is a fulminant hepatitis that is frequently fatal (see Dreifuss et al., 1989). Pathological examination reveals a microvesicular steatosis without evidence of inflammation or hypersensitivity reaction. Children below 2 years of age with other medical conditions who were given multiple antiseizure agents were especially likely to suffer fatal hepatic injury. At the other extreme, there were no deaths reported for patients over the age of 10 years who received only valproate. Acute pancreatitis and hyperammonemia also have been frequently associated with the use of valproic acid. Plasma Drug Concentrations The concentration of valproate in plasma that is associated with
therapeutic effects is approximately 30 to 100 Drug Interactions Valproate primarily inhibits drugs metabolized by CYP2C9 including phenytoin and phenobarbital. Valproate also inhibits UGT and thus inhibits the metabolism of lamotrigine and lorazepam. A high proportion of valproate is bound to albumin, and the high molar concentrations of valproate in the clinical setting result in valproate's displacing phenytoin and other drugs from albumin. With respect to phenytoin in particular, valproate's inhibition of the drug's metabolism is countered by displacement of phenytoin from albumin. The concurrent administration of valproate and clonazepam has been associated with the development of absence status epilepticus; however, this complication appears to be rare. Therapeutic Uses Valproate is effective in the treatment of absence, myoclonic, partial, and tonic-clonic seizures. The initial daily dose usually is 15 mg/kg, and this is increased at weekly intervals by 5 to 10 mg/kg per day to a maximum daily dose of 60 mg/kg. Divided doses should be given when the total daily dose exceeds 250 mg. The therapeutic uses of valproate in epilepsy are discussed further at the end of this chapter. |
Benzodiazepines
|
The benzodiazepines are employed clinically
primarily as sedative-antianxiety drugs; their pharmacology is presented in
detail in Chapters 17: Hypnotics and Sedatives and 19: Drugs and the
Treatment of Psychiatric Disorders: Depression and Anxiety Disorders.
Discussion in this chapter is limited to consideration of their usefulness in
the therapy of the epilepsies. A large number of benzodiazepines have broad
antiseizure properties, but only clonazepam (KLONOPIN) and clorazepate (TRANXENE-SD; others) have been approved in
the Antiseizure Properties In animals, prevention of pentylenetetrazol-induced seizures by the benzodiazepines is much more prominent than is their modification of the maximal electroshock seizure pattern. Clonazepam is unusually potent in antagonizing the effects of pentylenetetrazol, but it is almost without action on seizures induced by maximal electroshock. Benzodiazepines, including clonazepam, suppress the spread of kindled seizures and generalized convulsions produced by stimulation of the amygdala, but do not abolish the abnormal discharge at the site of stimulation. Mechanism of Action The antiseizure actions of the benzodiazepines, as well as other effects that occur at nonsedating doses, result in large part from their ability to enhance GABA-mediated synaptic inhibition. Molecular cloning and study of recombinant receptors have demonstrated that the benzodiazepine receptor is an integral part of the GABAA receptor (see Chapter 17: Hypnotics and Sedatives). At therapeutically relevant concentrations, benzodiazepines act at subsets of GABAA receptors and increase the frequency, but not duration, of openings at GABA-activated chloride channels (Twyman et al., 1989). At higher concentrations, diazepam and many other benzodiazepines can reduce sustained high-frequency firing of neurons, similar to the effects of phenytoin, carbamazepine, and valproate. Although these concentrations correspond to those achieved in patients during treatment of status epilepticus with diazepam, they are considerably higher than those associated with antiseizure or anxiolytic effects in ambulatory patients. Pharmacokinetic Properties Benzodiazepines are well absorbed after oral administration, and concentrations in plasma are usually maximal within 1 to 4 hours. After intravenous administration, they are redistributed in a manner typical of that for highly lipid-soluble agents (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination). Central effects develop promptly, but wane rapidly as the drugs move to other tissues. Diazepam is redistributed especially rapidly, with a half-life of redistribution of about 1 hour. The extent of binding of benzodiazepines to plasma proteins correlates with lipid solubility, ranging from approximately 99% for diazepam to about 85% for clonazepam (see Appendix II). The major metabolite of diazepam, N-desmethyl-diazepam, is somewhat less active than the parent drug and may behave as a partial agonist. This metabolite also is produced by the rapid decarboxylation of clorazepate following its ingestion. Both diazepam and N-desmethyl-diazepam are slowly hydroxylated to other active metabolites, such as oxazepam. The half-life of diazepam in plasma is between 1 and 2 days, while that of N-desmethyl-diazepam is about 60 hours. Clonazepam is metabolized principally by reduction of the nitro group to produce inactive 7-amino derivatives. Less than 1% of the drug is recovered unchanged in the urine. The half-life of clonazepam in plasma is about 1 day. Lorazepam is metabolized chiefly by conjugation with glucuronic acid; its half-life in plasma is about 14 hours. Toxicity The principal side effects of long-term oral therapy with clonazepam are drowsiness and lethargy. These occur in about 50% of patients initially, but tolerance often develops with continued administration. Muscular incoordination and ataxia are less frequent. Although these symptoms usually can be kept to tolerable levels by reducing the dosage or the rate at which it is increased, they sometimes force discontinuation of the drug. Other side effects include hypotonia, dysarthria, and dizziness. Behavioral disturbances, especially in children, can be very troublesome; these include aggression, hyperactivity, irritability, and difficulty in concentration. Both anorexia and hyperphagia have been reported. Increased salivary and bronchial secretions may cause difficulties in children. Seizures are sometimes exacerbated, and status epilepticus may be precipitated if the drug is discontinued abruptly. Other aspects of the toxicity of the benzodiazepines are discussed in Chapter 17: Hypnotics and Sedatives. Cardiovascular and respiratory depression may occur after the intravenous administration of diazepam, clonazepam, or lorazepam, particularly if other antiseizure agents or central depressants have been administered previously. Plasma Drug Concentrations Because tolerance affects the relationship between drug concentration and drug antiseizure effect, plasma concentrations of benzodiazepines are of limited value. Therapeutic Uses Clonazepam is useful in the therapy of absence seizures as well as myoclonic seizures in children. However, tolerance to its antiseizure effects usually develops after 1 to 6 months of administration, after which some patients no longer will respond to clonazepam at any dosage. The initial dose of clonazepam for adults should not exceed 1.5 mg per day and for children is 0.01 to 0.03 mg/kg per day. The dose-dependent side effects are reduced if two or three divided doses are given each day. The dose may be increased every 3 days in amounts of 0.25 to 0.5 mg per day in children and 0.5 to 1 mg per day in adults. The maximal recommended dose is 20 mg per day for adults and 0.2 mg/kg per day for children. While diazepam is an effective agent for treatment of status epilepticus, its short duration of action is a disadvantage, leading to the frequent use of intravenous phenytoin in combination with diazepam. Diazepam is administered intravenously and at a rate of no more than 5 mg per minute. The usual dose for adults is 5 to 10 mg, as required; this may be repeated at intervals of 10 to 15 minutes, up to a maximal dose of 30 mg. If necessary, this regimen can be repeated in 2 to 4 hours, but no more than 100 mg should be administered in a 24-hour period. Although diazepam is not useful as an oral agent for the treatment of seizure disorders, clorazepate is effective in combination with certain other drugs in the treatment of partial seizures. The maximal inital dose of clorazepate is 22.5 mg per day in three portions for adults and 15 mg per day in two doses in children. Clorazepate is not recommended for children under the age of 9. |
Other Antiseizure Agents
|
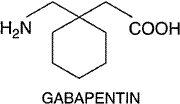
Gabapentin Gabapentin NEURONTIN) is an antiseizure drug that was approved by the United States Food and Drug Administration in 1993. The chemical structure of gabapentin is a GABA molecule covalently bound to a lipophilic cyclohexane ring. Gabapentin was designed to be a centrally active GABA agonist, its high lipid solubility aimed at facilitating its transfer across the blood-brain barrier. The structure of gabapentin is shown below:
Pharmacological Effects and Mechanisms of Action Gabapentin inhibits tonic hindlimb extension in the electroshock
seizure model. Interestingly, gabapentin also inhibits clonic seizures
induced by pentylenetetrazol. Its efficacy in both these tests parallels that
of valproic acid and distinguishes it from phenytoin and carbamazepine. The
anticonvulsant mechanism of action of gabapentin is unknown. Despite its
design as a GABA agonist, gabapentin does not mimic GABA when
iontophoretically applied to neurons in primary culture. Gabapentin may
promote nonvesicular release of GABA through a poorly understood mechanism (Honmou
et al., 1995). Gabapentin does bind a protein in cortical membranes
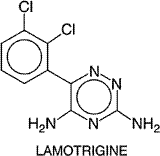
with an amino acid sequence identical to that of the Pharmacokinetics Gabapentin is absorbed after oral administration and is not metabolized in human beings. It is excreted unchanged, mainly in the urine. Its half-life, when it is used as monotherapy, is 5 to 9 hours. Concurrent administration of gabapentin does not affect the plasma concentrations of phenytoin, carbamazepine, phenobarbital, or valproate. Therapeutic Uses Gabapentin is approved by the FDA for treating partial seizures, with and without secondary generalization, in adults when used in addition to other antiseizure drugs. Double-blind, placebo-controlled trials of patients with refractory partial seizures demonstrated that addition of gabapentin to other antiseizure drugs was superior to placebo. The median seizure decrease induced by gabapentin was approximately 27% compared with 12% for placebo. A double-blind study of gabapentin (900 or 1800 mg/day) monotherapy disclosed that gabapentin was similar in efficacy to carbamazepine (600 mg/day). Gabapentin also is being used for migraine, chronic pain, and bipolar disorder. Gabapentin usually is effective in doses of 900 to 1800 mg daily in three doses. Therapy usually is begun with a low dose (300 mg once on the first day), and the dose is increased in daily increments of 300 mg until an effective dose is reached. Toxicity The most common adverse effects of gabapentin are somnolence, dizziness, ataxia, and fatigue. These effects usually are mild to moderate in severity but resolve within two weeks of onset during continued treatment. Overall, gabapentin is well tolerated. Lamotrigine Lamotrigine LAMICTAL) is a phenyltriazine derivative initially developed as an antifolate agent based upon the incorrect idea that reducing folate would effectively combat seizures. Structure-activity studies indicate that its effectiveness as an antiseizure drug is unrelated to its antifolate properties (Macdonald and Greenfield, 1997). It was approved by the Food and Drug Administration in 1994. Its chemical structure is:
Pharmacological Effects and Mechanisms of Action Lamotrigine suppresses tonic hindlimb extension in the maximal electroshock model and partial and secondarily generalized seizures in the kindling model but does not inhibit clonic motor seizures induced by pentylenetetrazol. Lamotrigine blocks sustained repetitive firing of mouse spinal cord neurons and delays the recovery from inactivation of recombinant Na+ channels, mechanisms similar to those of phenytoin and carbamazepine (Xie et al., 1995). This may well explain lamotrigine's actions on partial and secondarily generalized seizures. However, as mentioned below, lamotrigine is effective against a broader spectrum of seizures than phenytoin and carbamazepine, suggesting that lamotrigine may have actions in addition to regulating recovery from inactivation of Na+ channels. The mechanisms underlying its broad spectrum of actions are incompletely understood. One possibility involves lamotrigine's inhibition of glutamate release in rat cortical slices treated with veratridine, a Na+ channel activator, raising the possibility that lamotrigine inhibits synaptic release of glutamate by acting at Na+ channels themselves. Pharmacokinetics Lamotrigine is completely absorbed from the gastrointestinal tract and is metabolized primarily by glucuronidation. The plasma half-life of a single dose is 24 to 35 hours. Administration of phenytoin, carbamazepine, phenobarbital, or primidone reduces the half-life of lamotrigine to approximately 15 hours and reduces plasma concentrations of lamotrigine. Conversely, addition of valproate markedly increases plasma concentrations of lamotrigine, likely by inhibiting glucuronidation. Addition of lamotrigine to valproic acid produces a reduction of valproate concentrations by approximately 25% over a few weeks. Concurrent use of lamotrigine and carbamazepine is associated with increases of the 10,11-epoxide of carbamazepine and clinical toxicity in some patients. Therapeutic Use Lamotrigine is useful for monotherapy and add-on therapy of partial and secondarily generalized tonic-clonic seizures in adults and Lennox-Gastaut syndrome in both children and adults. A double-blind comparison of lamotrigine and carbamazepine monotherapy in newly diagnosed partial or generalized tonic-clonic seizures disclosed similar efficacy for the two drugs, but lamotrigine was better tolerated (Brodie et al., 1995). A double-blind, placebo-controlled trial of addition of lamotrigine to existing antiseizure drugs demonstrated effectiveness of lamotrigine against tonic-clonic seizures and drop attacks in children with the Lennox-Gastaut syndrome (Motte et al., 1997). Lennox-Gastaut syndrome is a disorder of childhood characterized by multiple seizure types, mental retardation, and refractoriness to antiseizure medication. There also is emerging evidence that lamotrigine is effective against juvenile myoclonic epilepsy and absence epilepsy. Patients who already are taking a hepatic enzyme-inducing antiseizure drug (such as carbamazepine, phenytoin, phenobarbital, or primidone, but not valproate) should be given lamotrigine initially at 50 mg per day for 2 weeks. The dose is increased to 50 mg twice per day for 2 weeks and then increased in increments of 100 mg/day each week up to a maintenance dose of 300 to 500 mg/day divided into two doses. For patients taking valproate in addition to an enzyme-inducing antiseizure drug, the initial dose should be 25 mg every other day for 2 weeks, followed by an increase to 25 mg/day for 2 weeks; the dose then can be increased by 25 to 50 mg/day every 1 to 2 weeks up to a maintenance dose of 100 to 150 mg/day divided into two doses. Toxicity The most common adverse effects are dizziness, ataxia, blurred or double vision, nausea, vomiting, and rash when lamotrigine was added to another antiseizure drug. A few cases of Stevens-Johnson syndrome and disseminated intravascular coagulation have been reported. Acetazolamide Acetazolamide, the prototype for the carbonic anhydrase inhibitors, is discussed in Chapter 29: Diuretics. Its antiseizure actions are discussed in previous editions of this textbook. Although it is sometimes effective against absence seizures, its usefulness is limited by the rapid development of tolerance. Adverse effects are minimal when it is used in moderate dosage for limited periods. Felbamate Felbamate FELBATOL) is a dicarbamate which was approved by the Food and Drug Administration for partial seizures in 1993. An association between felbamate and aplastic anemia in at least ten cases resulted in a recommendation by the Food and Drug Administration and the manufacturer for the immediate withdrawal of most patients from treatment with this drug. The structure of felbamate is shown below:
Felbamate is effective in both the maximal electroshock and pentylenetetrazol seizure models. Clinically relevant concentrations of felbamate inhibit NMDA-evoked responses and potentiate GABA-evoked responses in whole-cell, voltage-clamp recordings of cultured rat hippocampal neurons (Rho et al., 1994). This dual action on excitatory and inhibitory transmitter responses may contribute to the wide spectrum of action of the drug in seizure models. An active control, randomized, double-blind protocol demonstrated the efficacy of felbamate in patients with poorly controlled partial and secondarily generalized seizures (Sachdeo et al., 1992). Felbamate also was found to be efficacious against seizures in patients with Lennox-Gastaut syndrome (The Felbamate Study Group in Lennox-Gastaut Syndrome, 1993). The clinical efficacy of this compound, which inhibited responses to NMDA and potentiated those to GABA, underscores the potential value of additional antiseizure agents with similar mechanisms of action. Levetiracetam Levetiracetam KEPPRA) is a
pyrrolidine, the racemically pure S-enantiomer of
Pharmacological Effects and Mechanism of Action Levetiracetam exhibits a novel pharmacological profile insofar as it inhibits partial and secondarily generalized tonic-clonic seizures in the kindling model yet is ineffective against maximum electroshock- and pentylenetetrazol-induced seizures, findings consistent with effectiveness against partial and secondarily generalized tonic-clonic seizures clinically. The mechanism by which levetiracetam exerts these antiseizure effects is unknown. No evidence for an action on voltage-gated Na+ channels or either GABA-or glutamate-mediated synaptic transmission has emerged. A stereoselective binding site has been identified in rat brain membranes, but the molecular identity of this site remains obscure. Pharmacokinetics Levetiracetam is rapidly and almost completely absorbed after oral administration. Ninety-five percent of the drug and its metabolite are excreted in the urine, 65% of which is unchanged drug; 24% of the drug is metabolized by hydrolysis of the acetamide group. It neither induces nor is a high-affinity substrate for cytochrome P450 isoforms or glucuronidation enzymes and thus is devoid of known interactions with other antiseizure drugs, oral contraceptives, or anticoagulants. Therapeutic Use A double-blind, placebo-controlled trial of adults with refractory partial seizures demonstrated that addition of levetiracetam to other antiseizure medications was superior to placebo. Its efficacy for monotherapy is being investigated. Toxicity The drug is well tolerated. The most frequently reported adverse effects are somnolence, asthenia, and dizziness. Tiagabine Tiagabine GABITRIL) is a derivative of nipecotic acid that was approved by the Food and Drug Administration in 1998 for treating partial seizures in adults when used in addition to other drugs. Its structure is as follows:
Pharmacological Effects and Mechanism of Action Tiagabine inhibits the GABA transporter, GAT-1, and thereby reduces GABA uptake into neurons and glia. In CA1 neurons of the hippocampus, tiagabine increases the duration of inhibitory synaptic currents, findings consistent with prolonging the effect of GABA at inhibitory synapses through reducing its reuptake by GAT-1. Tiagabine inhibits maximum electroshock seizures and both limbic and secondarily generalized tonic-clonic seizures in the kindling model, results suggestive of efficacy against partial and tonic-clonic seizures clinically. Pharmacokinetics Tiagabine is rapidly absorbed after oral administration, extensively bound to proteins, and metabolized mainly in the liver and predominantly by CYP3A. Its half-life is about 8 hours but is shortened by 2 to 3 hours when coadministered with hepatic enzyme-inducing drugs such as phenobarbital, phenytoin, or carbamazepine. Therapeutic Use Double-blind, placebo-controlled trials have established tiagabine's efficacy as add-on therapy of refractory partial seizures with or without secondary generalization. Its efficacy for monotherapy for this indication has not yet been established. Toxicity The principal adverse effects include dizziness, somnolence, and tremor; they appear to be mild to moderate in severity, and appear shortly after drug initiation. The fact that tiagabine and other drugs thought to enhance effects of synaptically released GABA can facilitate spike-and-wave discharges in animal models of absence seizures raises the possibility that tiagabine may be contraindicated in patients with generalized absence epilepsy. Topiramate Topiramate TOPAMAX) is a sulfamate-substituted monosaccharide that was approved by the Food and Drug Administration in 1996 for partial seizures in adults when used in addition to other drugs. Its structure is as follows:
Pharmacological Effects and Mechanisms of Action Topiramate reduces voltage-gated Na+ currents in cerebellar granule cells and may act on the inactivated state of the channel in a manner similar to that of phenytoin. In addition, topiramate enhances postsynaptic GABAA-receptor currents and also limits activation of the AMPA-kainate-subtype(s) of glutamate receptor. Topiramate also is a weak carbonic anhydrase inhibitor. Topiramate inhibits maximal electroshock and pentylenetetrazol-induced seizures as well as partial and secondarily generalized tonic-clonic seizures in the kindling model, findings predictive of a broad spectrum of antiseizure actions clinically. Pharmacokinetics Topiramate is rapidly absorbed after oral administration and is mainly excreted unchanged in the urine. The remainder undergoes metabolism by hydroxylation, hydrolysis, and glucuronidation with no one metabolite accounting for more than 5% of an oral dose. Its half-life is about a day. Reduced estradiol plasma concentrations occur with concurrent topiramate, suggesting the need for higher doses of oral contraceptives when coadministered with topiramate. Therapeutic Use Double-blind, placebo-controlled studies established the efficacy of topiramate in both adults and children with refractory partial seizures with or without secondary generalized tonic-clonic seizures. Topiramate also was found to be significantly more effective than placebo against both drop attacks and tonic-clonic seizures in patients with Lennox-Gastaut syndrome and against tonic-clonic and myoclonic seizures in adults and children with primary generalized epilepsy. A pilot study suggests that topiramate may be effective against infantile spasms. Toxicity Topiramate is well tolerated. The most common adverse effects are somnolence, fatigue, weight loss, and nervousness. Zonisamide Zonisamide ZONEGRAN) is a sulfonamide derivative that was approved by the Food and Drug Administration in 2000 for partial seizures in adults when used in addition to other drugs. Its structure is as follows:
Pharmacological Effects and Mechanism of Action Zonisamide inhibits the T-type Ca2+ currents. In addition,
zonisamide inhibits the sustained, repetitive firing of spinal cord neurons,
presumably by prolonging the inactivated state of voltage-gated Na+
channels in a manner similar to actions of phenytoin and carbamazepine.
Zonisamide inhibits tonic hindlimb extension evoked by maximal electroshock
and inhibits both partial and secondarily generalized seizures in the
kindling model, results predictive of clinical effectiveness against partial
and secondarily generalized tonic-clonic seizures. Zonisamide does not
inhibit minimal clonic seizures induced by pentylenetetrazol, suggesting that
the drug will not be effective clinically against myoclonic seizures.
Zonisamide's inhibition of T-type Ca2+ currents suggests that it
may be effective against absence seizures, yet its effects in absence models
such as the lethargic mouse or the absence epileptic rat of Pharmacokinetics Zonisamide is almost completely absorbed after oral administration, has a long half-life (about 63 hours), and is about 40% bound to plasma protein. Approximately 85% of an oral dose is excreted in the urine, principally as unmetabolized zonisamide and a glucuronide of sulfamoylacetyl phenol, which is a product of metabolism by CYP3A4. Phenobarbital, phenytoin, and carbamazepine decrease the plasma concentration/dose ratio of zonisamide, whereas lamotrigine increases this ratio. Conversely, zonisamide has little effect on the plasma concentrations of other antiseizure drugs. Therapeutic Use Double-blind, placebo-controlled studies of patients with refractory partial seizures demonstrated that addition of zonisamide to other drugs was superior to placebo. Additional studies of zonisamide have been initiated in absence seizures, infantile spasms, and Lennox-Gastaut syndrome, but only largely anecdotal data are currently available. Toxicity Overall, zonisamide is well tolerated. The most common adverse effects include somnolence, ataxia, anorexia, nervousness, and fatigue. Approximately 1% of individuals develop renal calculi during treatment with zonisamide; the mechanism of this effect is obscure. |
General Principles and Choice of Drugs for the Therapy of the Epilepsies
|
Early diagnosis and treatment of seizure disorders with a single appropriate agent offers the best prospect of achieving prolonged seizure-free periods with the lowest risk of toxicity. An attempt should be made to determine the cause of the epilepsy with the hope of discovering a correctable lesion, either structural or metabolic. The efficacy of antiseizure drugs has been assessed in clinical trials on the basis of seizure type, not epilepsy syndrome type, and thus seizure type determines drug selection. The drugs commonly used for distinct seizure types are listed in Table 211. The efficacy combined with the unwanted effects of a given drug determine which particular drug is optimal for a given patient. The first issue that arises is whether or not and when to initiate treatment. For example, it may not be necessary to initiate antiseizure therapy after an isolated tonic-clonic seizure in a healthy young adult who lacks a family history of epilepsy and who has a normal neurological exam, a normal EEG, and a normal brain magnetic resonance imaging (MRI) scan. That is, the odds of seizure recurrence in the next year (15%) approximate the risk of a drug reaction sufficiently severe to warrant discontinuation of medication (Bazil and Pedley, 1998). Alternatively, a similar seizure occurring in an individual with a positive family history of epilepsy, an abnormal neurological exam, an abnormal EEG, and an abnormal MRI carries a risk of recurrence approximating 60%, odds that favor initiation of therapy. Unless extenuating circumstances such as status epilepticus exist, medication should be initiated with a single drug. Initial dosage usually is that expected to provide a plasma drug concentration during the plateau state at least in the lower portion of the range associated with clinical efficacy. To minimize dose-related adverse effects, therapy with many drugs is initiated at reduced dosage. Dosage is increased at appropriate intervals, as required for control of seizures or as limited by toxicity, and such adjustment is preferably assisted by monitoring of drug concentrations in plasma. Compliance with a properly selected, single drug in maximal tolerated dosage results in complete control of seizures in approximately 50% of patients. If a seizure occurs despite optimal drug levels, the physician should assess the presence of potential precipitating factors such as sleep deprivation, a concurrent febrile illness, or drugs; drugs might consist of large amounts of caffeine or even over-the-counter medications, which can include drugs that can lower the seizure threshold. If compliance has been confirmed yet seizures persist, another drug should be substituted. Unless serious adverse effects of the drug dictate otherwise, dosage always should be reduced gradually when a drug is being discontinued to minimize risk of seizure recurrence. In the case of partial seizures in adults, the diversity of available drugs permits selection of a second drug that acts by a distinct mechanism. Smith et al. (1987) found that 55% of such patients could be managed satisfactorily on a second single drug, yet others report that only 9% to 11% of patients with complex partial seizures failing an initial drug achieve complete seizure control with a second single drug (Schmidt and Richter, 1986; Dasheiff et al., 1986). In the event that therapy with a second single drug also is inadequate, many physicians resort to treatment with two drugs simultaneously. This decision should not be taken lightly, because most patients obtain optimal seizure control with fewest unwanted effects when taking a single drug. Nonetheless, some patients will not be controlled adequately without the use of two or more antiseizure agents simultaneously. No properly controlled studies have compared systematically one particular drug combination with another. It seems wise to select two drugs that act by distinct mechanisms (e.g., one that promotes Na+ channel inactivation and another that enhances GABA-mediated synaptic inhibition). Additional issues that warrant careful consideration are the unwanted effects of each drug and the potential drug interactions. As specified in Table 212, many of these drugs induce expression of cytochrome P450 enzymes and thereby impact the metabolism of themselves and/or other drugs. Overall, the more recently developed antiseizure drugs present fewer problems with respect to drug interactions. If a patient fails two drugs in monotherapy, the odds that polytherapy will provide complete control are small. Alternative measures such as epilepsy surgery should be considered. Essential to optimal management of epilepsy is the filling out of a seizure chart by the patient or a relative. Frequent visits to the physician or seizure clinic may be necessary early in the period of treatment, since hematological and other possible side effects may require consideration of a change in medication. Long-term follow-up with neurological examinations and possibly EEG and neuroimaging studies is appropriate. Most crucial for successful management is regularity of medication, since faulty compliance is the most frequent cause for failure of therapy with antiseizure drugs. Measurement of plasma drug concentration at appropriate intervals greatly facilitates the initial adjustment of dosage for individual differences in drug elimination and the subsequent adjustment of dosage to minimize dose-related adverse effects without sacrifice of seizure control. Periodic monitoring during maintenance therapy can detect failure of the patient to take the medication as prescribed. Knowledge of plasma drug concentration can be especially helpful during multiple-drug therapy. If toxicity occurs, monitoring helps to identify the particular drug(s) responsible, and if pharmacokinetic drug interaction occurs, monitoring can guide readjustment of dosage. Duration of Therapy In an attempt to provide guidelines for withdrawal of antiseizure drugs, Shinnar et al. (1994) prospectively studied 264 children in whom antiseizure drugs were discontinued after a mean seizure-free interval of 2.9 years. Children were followed for a mean of 58 months to assess seizure recurrence. Seizures recurred in 36% of children. Factors associated with an increased risk of recurrence included a positive family history of epilepsy, presence of slowing on EEG prior to withdrawal, onset of epilepsy after age 12 (compared with younger ages), atypical febrile seizures, and certain epileptic syndromes such as juvenile myoclonic epilepsy. In a prospective study, the treatment of patients with generalized or partial seizures was stopped after 2 seizure-free years; only patients who had been treated with a single drug (phenytoin, carbamazepine, or valproate) were included (Callaghan et al., 1988). The overall rate of relapse (within 3 years) was approximately 33% in both children and adults. Although only 92 patients were studied, the risk of relapse was apparently greatest for patients with complex partial seizures or those who had a persistently abnormal EEG. Although these and other results are encouraging, it is not yet possible to provide clear guidelines for the selection of patients for withdrawal from therapy. Such decisions must be made on an individual basis, weighing both the medical and psychosocial consequences of recurrence of seizures against the potential toxicity associated with prolonged therapy. If a decision to withdraw antiseizure drugs is made, such withdrawal should be done gradually over a period of months. The risk of status epilepticus is increased with abrupt cessation of therapy. Simple and Complex Partial and Secondarily Generalized Tonic-Clonic Seizures The efficacy and toxicity of carbamazepine, phenobarbital, phenytoin, and primidone for treatment of partial and secondarily generalized tonic-clonic seizures in adults have been examined in a double-blind prospective study (Mattson et al., 1985). A subsequent double-blind prospective study compared carbamazepine with valproate (Mattson et al., 1992). Carbamazepine and phenytoin were the most effective overall for single-drug therapy of partial or generalized tonic-clonic seizures. The choice between carbamazepine and phenytoin required assessment of toxic effects of drugs. Primidone was associated with greater incidence of toxicity early in the course of therapy, including nausea, dizziness, ataxia, and somnolence. Decreased libido and impotence were associated with all four drugs (carbamazepine 13%, phenobarbital 16%, phenytoin 11%, and primidone 22%), but significantly more commonly with primidone. The study comparing carbamazepine with valproate revealed that carbamazepine provided superior control of complex partial seizures. With respect to adverse effects, carbamazepine was more commonly associated with skin rash, but valproate was more commonly associated with tremor and weight gain. Overall, the data demonstrated that carbamazepine and phenytoin are preferable for treatment of partial seizures, but phenobarbital, valproic acid, and primidone are efficacious. A double-blind comparison of lamotrigine and carbamazepine disclosed similar efficacy of the two drugs, but lamotrigine was better tolerated (Brodie et al., 1995). Lamotrigine is used for monotherapy of partial and secondarily generalized tonic-clonic seizures. Multiple drugs recently were approved for add-on therapy of these seizures, including gabapentin, levetiracetam, tiagabine, topiramate, and zonisamide. Control of secondarily generalized tonic-clonic seizures did not differ significantly with carbamazepine, phenobarbital, phenytoin, or primidone (Mattson et al., 1985). Valproate was as effective as carbamazepine for control of secondarily generalized tonic-clonic seizures (Mattson et al., 1992). Since secondarily generalized tonic-clonic seizures usually coexist with partial seizures, carbamazepine, phenytoin, and lamotrigine are the first-line drugs for these conditions. Absence Seizures The best current data indicate that ethosuximide and valproate are equally effective in the treatment of absence seizures (see Mikati and Browne, 1988). Between 50% and 75% of newly diagnosed patients can be rendered free of seizures. In the event that tonic-clonic seizures are present or emerge during therapy, valproate is the agent of first choice. Emerging evidence suggests that lamotrigine is effective for absence seizures (Bazil and Pedley, 1998). Myoclonic Seizures Valproic acid is the drug of choice for myoclonic seizures in the syndrome of juvenile myoclonic epilepsy, in which myoclonic seizures often coexist with tonic-clonic and also absence seizures. Monotherapy with lamotrigine may be effective in some patients with juvenile myoclonic epilepsy in whom valproic acid proves unsatisfactory (Bazil and Pedley, 1998). Febrile Convulsions Two percent to 4% of children experience a convulsion associated with a febrile illness. From 25% to 33% of these children will have another febrile convulsion. Only 2% to 3% become epileptic in later years. This is a sixfold increase in risk compared with the general population. Several factors are associated with an increased risk of developing epilepsy: preexisting neurological disorder or developmental delay, a family history of epilepsy, or a complicated febrile seizure (i.e., the febrile seizure lasted more than 15 minutes, was one-sided, or was followed by a second seizure in the same day). If all of these risk factors are present, the risk of developing epilepsy is only 10%. Concern regarding the increased risk of developing epilepsy or other neurological sequelae led many physicians to prescribe antiseizure drugs prophylactically after a febrile seizure. Uncertainties regarding the efficacy of prophylaxis for reducing epilepsy combined with substantial side effects of phenobarbital prophylaxis (Farwell et al., 1990) argue against the use of chronic therapy for prophylactic purposes (Freeman, 1992). For children at high risk of developing recurrent febrile seizures and epilepsy, rectally administered diazepam at the time of fever may prevent recurrent seizures and avoid side effects of chronic therapy. Seizures in Infants and Young Children Infantile spasms with hypsarrhythmia are refractory to the usual
antiseizure agents; corticotropin or the adrenocorticosteroids are commonly
used. A randomized study found vigabatrin ( The Lennox-Gastaut syndrome is a severe form of epilepsy which usually begins in childhood and is characterized by cognitive impairments and multiple types of seizures including tonic-clonic, tonic, atonic, myoclonic, and atypical absence seizures. Addition of lamotrigine to other antiseizure drugs resulted in improved seizure control in comparison to placebo in a double-blind trial (Motte et al., 1997), demonstrating lamotrigine to be an effective and well-tolerated drug for this treatment-resistant form of epilepsy. Felbamate also was found to be effective for seizures in this syndrome, but the occasional occurrence of aplastic anemia has limited its use. Status Epilepticus and Other Convulsive Emergencies Status epilepticus is a neurological emergency. Mortality for adults approximates 20% (Lowenstein and Alldredge, 1998). The goal of treatment is rapid termination of behavioral and electrical seizure activity; the longer the episode of status epilepticus is untreated, the more difficult it is to control and the risk of permanent brain damage increases. Critical to the management is a clear plan, prompt treatment with effective drugs in adequate doses, and attention to hypoventilation and hypotension. Since hypoventilation may result from high doses of drugs used for treatment, it may be necessary to assist respiration temporarily. Drugs should be administered by intravenous route only. Because of slow and unreliable absorption, the intramuscular route has no place in treatment of status epilepticus. To assess the optimal initial drug regimen, a double-blind, multicenter trial compared four intravenous treatments: diazepam followed by phenytoin; lorazepam; phenobarbital; and phenytoin alone (Treiman et al., 1998). The treatments were shown to have similar efficacies, in that success rates ranged from 44% to 65%, but lorazepam alone was significantly better than phenytoin alone. No significant differences were found with respect to recurrences or adverse reactions. Antiseizure Therapy and Pregnancy Use of antiseizure drugs has diverse implications of great importance
for the health of women, issues considered in guidelines articulated by the The effectiveness of oral contraceptives appears to be reduced by concomitant use of antiseizure drugs. The failure rate of oral contraceptives is 3.1/100 years in women receiving antiseizure drugs compared to a rate of 0.7/100 years in nonepileptic women. One attractive explanation of the increased failure rate is the increased rate of oral contraceptive metabolism caused by antiseizure drugs that induce hepatic enzymes (see Table 212); particular caution is needed with any antiseizure drug that induces CYP3A4. The apparent teratogenic effects of antiseizure drugs add to the deleterious consequences of oral contraceptive failure. Epidemiological evidence suggests that antiseizure drugs have teratogenic effects. Infants of epileptic mothers are at twofold greater risk of major congenital malformations than offspring of nonepileptic mothers (4% to 8% compared to 2% to 4%). These malformations include congenital heart defects, neural tube defects, and others. Inferring causality from the associations found in large epidemiological studies with many uncontrolled variables can be hazardous, but a causal role for antiseizure drugs is suggested by association of congenital defects with higher concentrations of a drug or with polytherapy compared to monotherapy. Phenytoin, carbamazepine, valproate, and phenobarbital all have been associated with teratogenic effects. Whether or not the recently developed antiseizure drugs also will be associated with teratogenic effects awaits clinical experience with these agents. One consideration for a woman with epilepsy who wishes to become pregnant is a trial free of antiseizure drug; monotherapy with careful attention to drug levels is another alternative. Polytherapy with toxic levels should be avoided. Folate supplementation (0.4 mg/day) has been recommended by the United States Public Health Service for all women of childbearing age to reduce the likelihood of neural tube defects, and this is appropriate for epileptic women as well. Antiseizure drugs that induce cytochrome P450 enzymes have been associated with vitamin K deficiency in the newborn, which can result in a coagulopathy and intracerebral hemorrhage in the neonate. Treatment with vitamin K1, 10 mg/day during the last month of gestation, has been recommended for prophylaxis. |
Prospectus
|
Improved therapies for epilepsy are likely to emerge from several lines of investigation over the next decade: (1) Clinical experience and additional clinical trials with the recently approved antiseizure drugs should optimize their utilization for diverse forms of epilepsy. (2) Increased insight into genetic, cellular, and molecular mechanisms of epilepsy emerging from basic investigations should lead to the development of drugs acting by mechanisms distinct from currently available medications. (3) Insight into cellular and molecular mechanisms of epileptogenesis emerging from studies of animal models should lead to pharmacological prophylaxis of individuals at high risk of developing epilepsy. (4) Pharmacogenomic investigations should optimize selection of antiseizure drugs efficacious in a given individual and permit identification of individuals at high risk for devastating, idiosyncratic drug effects. For further discussion of the epilepsies and convulsive disorders, see
Chapter 348 in Harrison's Principles of Internal Medicine, 16th ed., |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3557
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved