| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Neurotransmission: The Autonomic and Somatic Motor Nervous Systems
Overview
|
The theory of neurohumoral transmission received direct experimental validation nearly a century ago (see von Euler, 1981), and extensive investigation during the ensuing years led to its general acceptance. Nerves transmit information across most synapses and neuroeffector junctions by means of specific chemical agents known as neurohumoral transmitters or, more simply, neurotransmitters. The actions of many drugs that affect smooth muscle, cardiac muscle, and gland cells can be understood and classified in terms of their mimicking or modifying the actions of the neurotransmitters released by the autonomic fibers at either ganglia or effector cells. Most of the general principles concerning the physiology and pharmacology of the peripheral autonomic nervous system and its effector organs also apply with certain modifications to the neuromuscular junction of skeletal muscle and to the central nervous system (CNS). In fact, the study of neurotransmission in the CNS has benefited greatly from the delineation of this process in the periphery (see Chapter 12: Neurotransmission and the Central Nervous System). In both the CNS and the periphery, a series of specializations have evolved to permit the synthesis, storage, release, metabolism, and recognition of transmitters. These specializations govern the actions of the principal autonomic transmitters acetylcholine and norepinephrine. Other neurotransmitters, including several peptides, purines, and nitric oxide, secondarily mediate autonomic function. A clear understanding of the anatomy and physiology of the autonomic nervous system is essential to a study of the pharmacology of the intervening drugs. The actions of an autonomic agent on various organs of the body often can be predicted if the responses to nerve impulses that reach the organs are known. This chapter covers the anatomy, biochemistry, and physiology of the autonomic and somatic motor nervous systems, with emphasis on sites of action of drugs that are discussed in Chapters 7: Muscarinic Receptor Agonists and Antagonists, 8: Anticholinesterase Agents, 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia, and 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists. |
Anatomy and General Functions of the Autonomic and Somatic Motor Nervous Systems
|
The autonomic nervous system, as delineated
by Differences between Autonomic and Somatic Nerves The efferent nerves of the involuntary system supply all innervated structures of the body except skeletal muscle, which is served by somatic nerves. The most distal synaptic junctions in the autonomic reflex arc occur in ganglia that are entirely outside the cerebrospinal axis. These ganglia are small but complex structures that contain axodendritic synapses between preganglionic and postganglionic neurons. Somatic nerves contain no peripheral ganglia, and the synapses are located entirely within the cerebrospinal axis. Many autonomic nerves form extensive peripheral plexuses, but such networks are absent from the somatic system. Whereas motor nerves to skeletal muscles are myelinated, postganglionic autonomic nerves generally are nonmyelinated. When the spinal efferent nerves are interrupted, the skeletal muscles they innervate lack myogenic tone, are paralyzed, and atrophy, whereas smooth muscles and glands generally show some level of spontaneous activity independent of intact innervation. Visceral Afferent Fibers The afferent fibers from visceral structures are the first link in the reflex arcs of the autonomic system. With certain exceptions, such as local axon reflexes, most visceral reflexes are mediated through the central nervous system (CNS). The afferent fibers are, for the most part, nonmyelinated and are carried into the cerebrospinal axis by the vagus, pelvic, splanchnic, and other autonomic nerves. For example, about four-fifths of the fibers in the vagus are sensory. Other autonomic afferents from blood vessels in skeletal muscles and from certain integumental structures are carried in somatic nerves. The cell bodies of visceral afferent fibers lie in the dorsal root ganglia of the spinal nerves and in the corresponding sensory ganglia of certain cranial nerves, such as the nodose ganglion of the vagus. The efferent link of the autonomic reflex arc is discussed in the following sections. The autonomic afferent fibers are concerned with the mediation of visceral sensation (including pain and referred pain); with vasomotor, respiratory, and viscerosomatic reflexes; and with the regulation of interrelated visceral activities. An example of an autonomic afferent system is that arising from the pressoreceptive endings in the carotid sinus and the aortic arch and from the chemoreceptor cells in the carotid and aortic bodies; this system is important in the reflex control of blood pressure, heart rate, and respiration, and its afferent fibers pass in the glossopharyngeal and vagus nerves to the medulla oblongata in the brainstem. The neurotransmitters that mediate transmission from sensory fibers have not been unequivocally characterized. However, substance P is present in afferent sensory fibers, in the dorsal root ganglia, and in the dorsal horn of the spinal cord, and this peptide is a leading candidate for the neurotransmitter that functions in the passage of nociceptive stimuli from the periphery to the spinal cord and higher structures. Other neuroactive peptides, including somatostatin, vasoactive intestinal polypeptide (VIP), and cholecystokinin, also have been found in sensory neurons (Lundburg, 1996; Hkfelt et al., 2000), and one or more such peptides may play a role in the transmission of afferent impulses from autonomic structures. Enkephalins, present in interneurons in the dorsal spinal cord (within an area termed the substantia gelatinosa), have antinociceptive effects that appear to be brought about by presynaptic and postsynaptic actions to inhibit the release of substance P and diminish the activity of cells that project from the spinal cord to higher centers in the CNS. The excitatory amino acids, glutamate and aspartate, also play major roles in transmission of sensory responses to the spinal cord. Central Autonomic Connections There probably are no purely autonomic or somatic centers of integration, and extensive overlap occurs. Somatic responses always are accompanied by visceral responses and vice versa. Autonomic reflexes can be elicited at the level of the spinal cord. They clearly are demonstrable in the spinal animal, including human beings, and are manifested by sweating, blood pressure alterations, vasomotor responses to temperature changes, and reflex emptying of the urinary bladder, rectum, and seminal vesicles. Extensive central ramifications of the autonomic nervous system exist above the level of the spinal cord. For example, the integration of the control of respiration in the medulla oblongata is well known. The hypothalamus and the nucleus of the solitary tract (nucleus tractus solitarius) generally are regarded as principal loci of integration of autonomic nervous system functions, which include regulation of body temperature, water balance, carbohydrate and fat metabolism, blood pressure, emotions, sleep, respiration, and sexual responses. Signals are received through ascending spinobulbar pathways. Also, these areas receive input from the limbic system, neostriatum, cortex, and, to a lesser extent, other higher brain centers. Stimulation of the nucleus of the solitary tract and the hypothalamus activates bulbospinal pathways and hormonal output to mediate autonomic and motor responses in the organism (Andresen and Kunze, 1994; Loewy and Spyer, 1990; see also Chapter 12: Neurotransmission and the Central Nervous System). The hypothalamic nuclei that lie posteriorly and laterally are sympathetic in their main connections, while parasympathetic functions evidently are integrated by the midline nuclei in the region of the tuber cinereum and by nuclei lying anteriorly. Divisions of the Peripheral Autonomic System On the efferent side, the autonomic nervous system consists of two large divisions: (1) the sympathetic or thoracolumbar outflow and (2) the parasympathetic or craniosacral outflow. A brief outline of those anatomical features necessary for an understanding of the actions of autonomic drugs is given here. The arrangement of the principal parts of the peripheral autonomic nervous system is presented schematically in Figure 61. As discussed below, the neurotransmitter of all preganglionic autonomic fibers, all postganglionic parasympathetic fibers, and a few postganglionic sympathetic fibers is acetylcholine (ACh); these so-called cholinergic fibers are depicted in blue. The adrenergic fibers, shown in red, compose the majority of the postganglionic sympathetic fibers; here the transmitter is norepinephrine (noradrenaline, levarterenol). The terms cholinergic and adrenergic were proposed originally by Dale (1954) to describe neurons that liberate ACh and norepinephrine, respectively. As noted above, all of the transmitter(s) of the primary afferent fibers, shown in green, have not been identified conclusively. Substance P and glutamate are thought to mediate many afferent impulses; both are present in high concentrations in the dorsal regions of the spinal cord.
Sympathetic Nervous System The cells that give rise to the preganglionic fibers of this division lie mainly in the intermediolateral columns of the spinal cord and extend from the first thoracic to the second or third lumbar segment. The axons from these cells are carried in the anterior (ventral) nerve roots and synapse with neurons lying in sympathetic ganglia outside the cerebrospinal axis. The sympathetic ganglia are found in three locations: paravertebral, prevertebral, and terminal. The paravertebral sympathetic ganglia consist of 22 pairs that lie on either side of the vertebral column to form the lateral chains. The ganglia are connected to each other by nerve trunks and to the spinal nerves by rami communicantes. The white rami are restricted to the segments of the thoracolumbar outflow; they carry the preganglionic myelinated fibers that exit from the spinal cord by way of the anterior spinal roots. The gray rami arise from the ganglia and carry postganglionic fibers back to the spinal nerves for distribution to sweat glands and pilomotor muscles and to blood vessels of skeletal muscle and skin. The prevertebral ganglia lie in the abdomen and the pelvis near the ventral surface of the bony vertebral column and consist mainly of the celiac (solar), superior mesenteric, aorticorenal, and inferior mesenteric ganglia. The terminal ganglia are few in number, lie near the organs they innervate, and include ganglia connected with the urinary bladder and rectum and the cervical ganglia in the region of the neck. In addition, there are small intermediate ganglia, especially in the thoracolumbar region, that lie outside the conventional vertebral chain. They are variable in number and location but usually are in close proximity to the communicating rami and to the anterior spinal nerve roots. Preganglionic fibers issuing from the spinal cord may synapse with the neurons of more than one sympathetic ganglion. Their principal ganglia of termination need not correspond to the original level from which the preganglionic fiber exits the spinal cord. Many of the preganglionic fibers from the fifth to the last thoracic segment pass through the paravertebral ganglia to form the splanchnic nerves. Most of the splanchnic nerve fibers do not synapse until they reach the celiac ganglion; others directly innervate the adrenal medulla (see below). Postganglionic fibers arising from sympathetic ganglia innervate visceral structures of the thorax, abdomen, head, and neck. The trunk and the limbs are supplied by means of sympathetic fibers in spinal nerves, as previously described. The prevertebral ganglia contain cell bodies, the axons of which innervate the glands and the smooth muscles of the abdominal and the pelvic viscera. Many of the upper thoracic sympathetic fibers from the vertebral ganglia form terminal plexuses, such as the cardiac, esophageal, and pulmonary plexuses. The sympathetic distribution to the head and the neck (vasomotor, pupillodilator, secretory, and pilomotor) is by way of the cervical sympathetic chain and its three ganglia. All postganglionic fibers in this chain arise from cell bodies located in these three ganglia; all preganglionic fibers arise from the upper thoracic segments of the spinal cord, there being no sympathetic fibers that leave the CNS above the first thoracic level. The adrenal medulla and other chromaffin tissue are embryologically and anatomically similar to sympathetic ganglia; all are derived from the neural crest. The adrenal medulla differs from sympathetic ganglia in that the principal catecholamine that is released in human beings and many other species is epinephrine (adrenaline), whereas norepinephrine is released from postganglionic sympathetic fibers. The chromaffin cells in the adrenal medulla are innervated by typical preganglionic fibers that release acetylcholine. Parasympathetic Nervous System The parasympathetic nervous system consists of preganglionic fibers that originate in three areas of the CNS and their postganglionic connections. The regions of central origin are the midbrain, the medulla oblongata, and the sacral part of the spinal cord. The midbrain, or tectal, outflow consists of fibers arising from the Edinger-Westphal nucleus of the third cranial nerve and going to the ciliary ganglion in the orbit. The medullary outflow consists of the parasympathetic components of the seventh, ninth, and tenth cranial nerves. The fibers in the seventh cranial, or facial, nerve form the chorda tympani, which innervates the ganglia lying on the submaxillary and sublingual glands. They also form the greater superficial petrosal nerve, which innervates the sphenopalatine ganglion. The ninth cranial, or glossopharyngeal, autonomic components innervate the otic ganglion. Postganglionic parasympathetic fibers from these ganglia supply the sphincter of the iris (pupillae constrictor muscle), the ciliary muscle, the salivary and lacrimal glands, and the mucous glands of the nose, mouth, and pharynx. These fibers also include vasodilator nerves to the organs mentioned. The tenth cranial, or vagus, nerve arises in the medulla and contains preganglionic fibers, most of which do not synapse until they reach the many small ganglia lying directly on or in the viscera of the thorax and abdomen. In the intestinal wall, the vagal fibers terminate around ganglion cells in the plexuses of Auerbach and Meissner. Preganglionic fibers are thus very long, whereas postganglionic fibers are very short. The vagus nerve, in addition, carries a far greater number of afferent fibers (but apparently no pain fibers) from the viscera into the medulla; the cell bodies of these fibers lie mainly in the nodose ganglion. The parasympathetic sacral outflow consists of axons that arise from cells in the second, third, and fourth segments of the sacral cord and proceed as preganglionic fibers to form the pelvic nerves (nervi erigentes). They synapse in terminal ganglia lying near or within the bladder, rectum, and sexual organs. The vagal and sacral outflows provide motor and secretory fibers to thoracic, abdominal, and pelvic organs, as indicated in Figure 61. Enteric Nervous System Stimulation of particular vagal nuclei in the medulla oblongata or certain fibers in the vagal trunk was known for some time to elicit muscle relaxation in certain regions of the stomach or intestine, such as sphincters, instead of the expected and more common contractile response. In the mid-1960s, it became evident that relaxation of the gastrointestinal tract and other visceral organs was not necessarily mediated by adrenergic stimulation; rather, release of other putative transmitters from enteric neurons, located in Auerbach's and Meissner's plexuses, gave rise to hyperpolarization and relaxation of the smooth muscle (Figure 61). Over the succeeding years, certain peptides (i.e., VIP), nucleotides (ATP), and nitric oxide (NO) were found to be inhibitory transmitters in the gastrointestinal tract and other visceral organs (see Bennett, 1997). Inhibition is achieved either through guanylyl cyclase activation by nitric oxide or hyperpolarization through the activation of K+ channels. Specific K+ channel inhibitors such as apamin or inhibitors of nitric oxide synthase can distinguish the inhibitory events and their durations. Noncholinergic excitatory transmitters such as tachykinins (e.g., substance P) also are found to be released in regions of the enteric plexus. Substance P is a transmitter of the sensory afferent system, which is released locally or from afferent nerve branches that link to intramural ganglia. The enteric system does not have a unique connection to the CNS. While under the influence of parasympathetic preganglionic nerves, release of transmitters usually is dominated by local control. Coordination of contraction and relaxation at a local level would be expected for regulation of peristaltic waves in the intestine. Differences among Sympathetic, Parasympathetic, and Motor Nerves The sympathetic system is distributed to effectors throughout the body, whereas parasympathetic distribution is much more limited. Furthermore, the sympathetic fibers ramify to a much greater extent. A preganglionic sympathetic fiber may traverse a considerable distance of the sympathetic chain and pass through several ganglia before it finally synapses with a postganglionic neuron; also, its terminals make contact with a large number of postganglionic neurons. In some ganglia, the ratio of preganglionic axons to ganglion cells may be 1:20 or more. In this manner, a diffuse discharge of the sympathetic system is possible. In addition, synaptic innervation overlaps, so that one ganglion cell may be supplied by several preganglionic fibers. The parasympathetic system, in contrast, has its terminal ganglia very near to or within the organs innervated and thus is more circumscribed in its influences. In some organs a 1:1 relationship between the number of preganglionic and postganglionic fibers has been suggested, but the ratio of preganglionic vagal fibers to ganglion cells in Auerbach's plexus has been estimated as 1:8000. Hence, this distinction between the two systems does not apply to all sites. The cell bodies of somatic motor neurons are in the ventral horn of the spinal cord; the axon divides into many branches, each of which innervates a single muscle fiber, so that more than 100 muscle fibers may be supplied by one motor neuron to form a motor unit. At each neuromuscular junction, the axonal terminal loses its myelin sheath and forms a terminal arborization that lies in apposition to a specialized surface of the muscle membrane, termed the motor end-plate. Mitochondria and a collection of synaptic vesicles are concentrated at the nerve terminal. Through trophic influences of the nerve, those cell nuclei in the multinucleated skeletal muscle cell lying in close apposition to the synapse acquire the capacity to activate specific genes which express synapse-localized proteins (Hall and Sanes, 1993; Sanes and Lichtman, 1999). Details of Innervation The terminations of the postganglionic autonomic fibers in smooth muscle and glands form a rich plexus, or terminal reticulum. The terminal reticulum (sometimes called the autonomic ground plexus) consists of the final ramifications of the postganglionic sympathetic (adrenergic), parasympathetic (cholinergic), and visceral afferent fibers, all of which are enclosed within a frequently interrupted sheath of satellite or Schwann cells. At these interruptions, varicosities packed with vesicles are seen in the efferent fibers. Such varicosities occur repeatedly but at variable distances along the course of the ramifications of the axon. 'Protoplasmic bridges' occur between the smooth muscle fibers themselves at points of contact between their plasma membranes. They are believed to permit the direct conduction of impulses from cell to cell without the need for chemical transmission. These structures have been termed nexuses or tight junctions, and they enable the smooth muscle fibers to function as a unit or syncytium. Sympathetic ganglia are extremely complex, both anatomically and
pharmacologically (see Chapter 9: Agents Acting at the Neuromuscular
Junction and Autonomic Ganglia). The preganglionic fibers lose their myelin
sheaths and divide repeatedly into a vast number of end fibers with diameters
ranging from 0.1 to 0.3 Responses of Effector Organs to Autonomic Nerve Impulses From the responses of the various effector organs to autonomic nerve impulses and the knowledge of the intrinsic autonomic tone, one can predict the actions of drugs that mimic or inhibit the actions of these nerves. In most instances, the sympathetic and parasympathetic neurotransmitters can be viewed as physiological or functional antagonists. If one neurotransmitter inhibits a certain function, the other usually augments that function. Most viscera are innervated by both divisions of the autonomic nervous system, and the level of activity at any one moment represents the integration of influences of the two components. Despite the conventional concept of antagonism between the two portions of the autonomic nervous system, their activities on specific structures may be either discrete and independent or integrated and interdependent. For example, the effects of sympathetic and parasympathetic stimulation of the heart and the iris show a pattern of functional antagonism in controlling heart rate and pupillary aperture, respectively. Their actions on male sexual organs are complementary and are integrated to promote sexual function. The control of peripheral vascular resistance is primarily, but not exclusively, due to sympathetic control of arteriolar resistance. The effects of stimulating the sympathetic (adrenergic) and parasympathetic (cholinergic) nerves to various organs, visceral structures, and effector cells are summarized in Table 61. General Functions of the Autonomic Nervous System The integrating action of the autonomic nervous system is of vital
importance for the well-being of the organism. In general, the autonomic
nervous system regulates the activities of structures that are not under
voluntary control and that function below the level of consciousness. Thus,
respiration, circulation, digestion, body temperature, metabolism, sweating,
and the secretions of certain endocrine glands are regulated, in part or
entirely, by the autonomic nervous system. As Claude Bernard (18781879),
J.N. The sympathetic system and its associated adrenal medulla are not essential to life in a controlled environment. Under circumstances of stress, however, the lack of the sympathoadrenal functions becomes evident. Body temperature cannot be regulated when environmental temperature varies; the concentration of glucose in blood does not rise in response to urgent need; compensatory vascular responses to hemorrhage, oxygen deprivation, excitement, and exercise are lacking; resistance to fatigue is lessened; sympathetic components of instinctive reactions to the external environment are lost; and other serious deficiencies in the protective forces of the body are discernible. The sympathetic system normally is continuously active; the degree of activity varies from moment to moment and from organ to organ. In this manner, adjustments to a constantly changing environment are accomplished. The sympathoadrenal system also can discharge as a unit. This occurs particularly during rage and fright, when sympathetically innervated structures over the entire body are affected simultaneously. Heart rate is accelerated; blood pressure rises; red blood cells are poured into the circulation from the spleen (in certain species); blood flow is shifted from the skin and splanchnic region to the skeletal muscles; blood glucose rises; the bronchioles and pupils dilate; and, on the whole, the organism is better prepared for 'fight or flight.' Many of these effects result primarily from, or are reinforced by, the actions of epinephrine, secreted by the adrenal medulla (see below). In addition, signals are received in higher brain centers to facilitate purposeful responses or to imprint the event in memory. The parasympathetic system is organized mainly for discrete and localized discharge. Although it is concerned primarily with conservation of energy and maintenance of organ function during periods of minimal activity, its elimination is not compatible with life. Sectioning the vagus, for example, soon gives rise to pulmonary infection because of the inability of cilia to remove irritant substances from the respiratory tract. The parasympathetic system slows the heart rate, lowers the blood pressure, stimulates gastrointestinal movements and secretions, aids absorption of nutrients, protects the retina from excessive light, and empties the urinary bladder and rectum. Many parasympathetic responses are rapid and reflexive in nature. |
Neurotransmission
|
Nerve impulses elicit responses in smooth, cardiac, and skeletal muscles, exocrine glands, and postsynaptic neurons through liberation of specific chemical neurotransmitters. The steps involved and the evidence for them are presented in some detail because the concept of chemical mediation of nerve impulses profoundly affects our knowledge of the mechanism of action of drugs at these sites. Historical Aspects The earliest concrete proposal of a neurohumoral mechanism was made shortly
after the turn of the twentieth century. Lewandowsky (1898) and Langley
(1901) noted independently the similarity between the effects of injection of
extracts of the adrenal gland and stimulation of sympathetic nerves. A few
years later, in 1905, T.R. Elliott, while a student with Langley at
Cambridge, England, extended these observations and postulated that sympathetic
nerve impulses release minute amounts of an epinephrine-like substance in
immediate contact with effector cells. He considered this substance to be the
chemical step in the process of transmission. He also noted that, long after
sympathetic nerves had degenerated, the effector organs still responded
characteristically to the hormone of the adrenal medulla. In 1905, The studies of Otto Loewi, begun in 1921, provided the first direct evidence for the chemical mediation of nerve impulses by the release of specific chemical agents. Loewi stimulated the vagus nerve of a perfused (donor) frog heart and allowed the perfusion fluid to come in contact with a second (recipient) frog heart used as a test object. The recipient frog heart was found to respond, after a short lag, in the same way as did the donor heart. It was thus evident that a substance was liberated from the first organ that slowed the rate of the second. Loewi referred to this chemical substance as Vagusstoff ('vagus substance'; parasympathin); subsequently, Loewi and Navratil (1926) presented evidence to identify it as ACh. Loewi also discovered that an accelerator substance similar to epinephrine and called Acceleranstoff was liberated into the perfusion fluid in summer, when the action of the sympathetic fibers in the frog's vagus, a mixed nerve, predominated over that of the inhibitory fibers. Loewi's discoveries eventually were confirmed and became universally accepted. Evidence that the cardiac vagus-substance also is ACh in mammals was obtained in 1933 by Feldberg and Krayer. In addition to the role of ACh as the transmitter of all postganglionic parasympathetic fibers and of a few postganglionic sympathetic fibers, this substance has been shown to have transmitter function in three additional classes of nerves: preganglionic fibers of both the sympathetic and the parasympathetic systems, motor nerves to skeletal muscle, and certain neurons within the CNS. In the same year as Loewi's discovery, Cannon and Uridil (1921) reported that stimulation of the sympathetic hepatic nerves resulted in the release of an epinephrine-like substance that increased blood pressure and heart rate. Subsequent experiments firmly established that this substance is the chemical mediator liberated by sympathetic nerve impulses at neuroeffector junctions. Cannon called this substance 'sympathin.' In many of its pharmacological and chemical properties, 'sympathin' closely resembled epinephrine, but also differed in certain important respects. As early as 1910, Barger and Dale noted that the effects of sympathetic nerve stimulation were more closely reproduced by the injection of sympathomimetic primary amines than by that of epinephrine or other secondary amines. The possibility that demethylated epinephrine (norepinephrine) might be 'sympathin' had been repeatedly advanced, but definitive evidence for its being the sympathetic nerve mediator was not obtained until specific assays were developed for the determination of sympathomimetic amines in extracts of tissues and body fluids. von Euler in 1946 found that the sympathomimetic substance in highly purified extracts of bovine splenic nerve resembled norepinephrine by all criteria used. Norepinephrine is the predominant sympathomimetic substance in the postganglionic sympathetic nerves of mammals and is the adrenergic mediator liberated by their stimulation (see von Euler, 1972). Norepinephrine, its immediate precursor, dopamine, and epinephrine also are neurotransmitters in the CNS (see Chapter 12: Neurotransmission and the Central Nervous System). Evidence for Neurohumoral Transmission The concept of neurohumoral transmission or chemical neurotransmission was developed primarily to explain observations relating to the transmission of impulses from postganglionic autonomic fibers to effector cells. The general lines of evidence to support the concept have included (1) demonstration of the presence of a physiologically active compound and its biosynthetic enzymes at appropriate sites; (2) recovery of the compound from the perfusate of an innervated structure during periods of nerve stimulation but not (or in greatly reduced amounts) in the absence of stimulation; (3) demonstration that the compound is capable of producing responses identical with responses to nerve stimulation; and (4) demonstration that the responses to nerve stimulation and to the administered compound are modified in the same manner by various drugs, usually competitive antagonists. Chemical, rather than electrogenic, transmission at autonomic ganglia and the neuromuscular junction of skeletal muscle was not generally accepted for a considerable period, because techniques were limited in time and chemical resolution. Techniques of intracellular recording and microiontophoretic application of drugs as well as sensitive analytical assays have overcome these limitations. Neurotransmission in the peripheral and central nervous systems once was believed to conform to the hypothesis that each neuron contains only one transmitter substance. However, peptides, such as enkephalin, substance P, neuropeptide Y, VIP, and somatostatin; purines such as ATP or adenosine; and small molecules such as nitric oxide, have been found in nerve endings. These substances can depolarize or hyperpolarize nerve terminals or postsynaptic cells. Furthermore, results of histochemical, immunocytochemical, and autoradiographic studies have demonstrated that one or more of these substances is present in the same neurons that contain one of the classical biogenic amine neurotransmitters (Bartfai et al., 1988; Lundberg, 1996). For example, enkephalins are found in postganglionic sympathetic neurons and adrenal medullary chromaffin cells. VIP is localized selectively in peripheral cholinergic neurons that innervate exocrine glands, and neuropeptide Y is found in sympathetic nerve endings. These observations suggest that in many instances synaptic transmission may be mediated by the release of more than one neurotransmitter (see below). Steps Involved in Neurotransmission The sequence of events involved in neurotransmission is of particular importance pharmacologically, since the actions of most drugs modulate the individual steps. The term conduction is reserved for the passage of an impulse along an axon or muscle fiber; transmission refers to the passage of an impulse across a synaptic or neuroeffector junction. With the exception of the local anesthetics, very few drugs modify axonal conduction in the doses employed therapeutically. Hence, this process is described only briefly. Axonal Conduction Current knowledge of axonal conduction stems largely from the investigative work of Hodgkin and Huxley (1952). At rest, the interior of the typical mammalian axon is approximately 70 mV negative to the exterior. The resting potential is essentially a diffusion potential, based chiefly on the fortyfold higher concentration of K+ in the axoplasm as compared with the extracellular fluid and the relatively high permeability of the resting axonal membrane to this ion. Na+ and Cl are present in higher concentrations in the extracellular fluid than in the axoplasm, but the axonal membrane at rest is considerably less permeable to these ions; hence their contribution to the resting potential is small. These ionic gradients are maintained by an energy-dependent active transport or pump mechanism, which involves an adenosine triphosphatase (ATPase) activated by Na+ at the inner and by K+ at the outer surface of the membrane (see Hille, 1992; Hille et al., 1999a). In response to depolarization to a threshold level, an action potential or nerve impulse is initiated at a local region of the membrane. The action potential consists of two phases. Following a small gating current resulting from depolarization inducing an open conformation of the channel, the initial phase is caused by a rapid increase in the permeability of Na+ through voltage-sensitive Na+ channels. The result is inward movement of Na+ and a rapid depolarization from the resting potential, which continues to a positive overshoot. The second phase results from the rapid inactivation of the Na+ channel and the delayed opening of a K+ channel, which permits outward movement of K+ to terminate the depolarization. Inactivation appears to involve a voltage-sensitive conformational change in which a hydrophobic peptide loop physically occludes the open channel at the cytoplasmic side. Although not important in axonal conduction, Ca2+ channels in other tissues (e.g., heart) contribute to the action potential by prolonging depolarization by an inward movement of Ca2+. This influx of Ca2+ also serves as a stimulus to initiate intracellular events (Hille, 1992; Catterall, 2000). The transmembrane ionic currents produce local circuit currents around the axon. As a result of such localized changes in membrane potential, adjacent resting channels in the axon are activated, and excitation of an adjacent portion of the axonal membrane occurs. This brings about the propagation of the action potential without decrement along the axon. The region that has undergone depolarization remains momentarily in a refractory state. In myelinated fibers, permeability changes occur only at the nodes of Ranvier, thus causing a rapidly progressing type of jumping, or saltatory, conduction. The puffer fish poison, tetrodotoxin, and a close congener found in some shellfish, saxitoxin, selectively block axonal conduction; they do so by blocking the voltage-sensitive Na+ channel and preventing the increase in permeability to Na+ associated with the rising phase of the action potential. In contrast, batrachotoxin, an extremely potent steroidal alkaloid secreted by a South American frog, produces paralysis through a selective increase in permeability of the Na+ channel to Na+, which induces a persistent depolarization. Scorpion toxins are peptides that also cause persistent depolarization, but they do so by inhibiting the inactivation process (see Catterall, 2000). Na+ and Ca2+ channels are discussed in more detail in Chapters 15: Local Anesthetics, 32, and 35. Junctional Transmission The arrival of the action potential at the axonal terminals initiates a series of events that trigger transmission of an excitatory or inhibitory impulse across the synapse or neuroeffector junction. These events, diagrammed in Figure 62, are as follows.
Cholinergic Transmission Two enzymes, choline acetyltransferase and AChE, are involved in the synthesis and degradation, respectively, of ACh. Choline Acetyltransferase Choline acetyltransferase catalyzes the final step in the synthesis of AChthe acetylation of choline with acetyl coenzyme A (CoA; see Wu and Hersh, 1994; Parsons et al., 1993). The primary structure of choline acetyltransferase is known from molecular cloning, and its immunocytochemical localization has proven useful for identification of cholinergic axons and nerve cell bodies. Acetyl CoA for this reaction is derived from pyruvate via the multistep pyruvate dehydrogenase reaction or is synthesized by acetate thiokinase, which catalyzes the reaction of acetate with adenosine triphosphate (ATP) to form an enzyme-bound acyladenylate (acetyl AMP). In the presence of CoA, transacetylation and synthesis of acetyl CoA proceed. Choline acetyltransferase, like other protein constituents of the neuron, is synthesized within the perikaryon and then is transported along the length of the axon to its terminal. Axonal terminals contain a large number of mitochondria, where acetyl CoA is synthesized. Choline is taken up from the extracellular fluid into the axoplasm by active transport. The final step in the synthesis occurs within the cytoplasm, following which most of the ACh is sequestered within the synaptic vesicles. Although moderately potent inhibitors of choline acetyltransferase exist, they have no therapeutic utility, in part because the uptake of choline is the rate-limiting step in the biosynthesis of ACh. Choline Transport Transport of choline from the plasma into neurons is accomplished by
distinct high- and low-affinity transport systems. The high-affinity system
(Km= 1 to 5 Upon acetylation of choline, ACh is transported into and packaged in the synaptic vesicle. The vesicular transporter relies on a proton gradient to drive amine uptake. Vesamicol blocks ACh vesicular transport at micromolar concentrations. The genes for choline acetyltransferase and the vesicular transporter are found at the same locus, with the transporter gene positioned in the first intron of the transferase gene. Hence, a common promoter regulates the expression of both genes (Eiden, 1998). Acetylcholinesterase (AChE) For ACh to serve as a neurotransmitter in the motor system and certain neuronal synapses, it must be removed or inactivated within the time limits imposed by the response characteristics of the synapse. At the neuromuscular junction, immediate removal is required to prevent lateral diffusion and sequential activation of receptorswith 'flashlike suddenness,' as Dale expressed it. Modern biophysical methods have revealed that the time required for hydrolysis of ACh is less than a millisecond at the neuromuscular junction. Choline has only 103 to 105 of the potency of ACh at the neuromuscular junction. While AChE is found in cholinergic neurons (dendrites, perikarya, and axons), it is more widely distributed than cholinergic synapses. It is highly concentrated at the postsynaptic end-plate of the neuromuscular junction. Butyrylcholinesterase (BuChE; also known as pseudocholinesterase) is present in low abundance in glial or satellite cells but is virtually absent in neuronal elements of the central and peripheral nervous systems. BuChE is synthesized primarily in the liver and is found in liver and plasma; its likely vestigial physiological function is the hydrolysis of ingested esters from plant sources. AChE and BuChE typically are distinguished by the relative rates of ACh and butyrylcholine hydrolysis and by effects of selective inhibitors (see Chapter 8: Anticholinesterase Agents). Almost all the pharmacological effects of the anti-ChE agents are due to the inhibition of AChE, with the consequent accumulation of endogenous ACh in the vicinity of the nerve terminal. Distinct, but single, genes encode AChE and BuChE in mammals; the diversity of molecular structures of AChE arise from alternative mRNA processing (Taylor et al., 2000). Storage and Release of Acetylcholine Fatt and Katz (1952) recorded at the motor end-plate of skeletal muscle and observed the random occurrence of small (0.1 to 3.0 mV), spontaneous depolarizations at a frequency of approximately one per second. The magnitude of these miniature end-plate potentials (mepps) is considerably below the threshold required to fire a muscle AP; that they are due to the release of ACh is indicated by their enhancement by neostigmine (an anti-ChE agent) and their blockade by d-tubocurarine (a competitive antagonist that acts at nicotinic receptors). These results led to the hypothesis that ACh is released from motor-nerve endings in constant amounts, or quanta. The likely morphological counterpart of quantal release was discovered shortly thereafter in the form of synaptic vesicles (De Robertis and Bennett, 1955). Most of the storage and release properties of ACh originally investigated in motor end-plates apply to other fast-responding synapses. When an action potential arrives at the motor-nerve terminal, there is a synchronous release of 100 or more quanta (or vesicles) of ACh (Katz and Miledi, 1965). Estimates of the ACh content of synaptic vesicles range from 1000 to over 50,000 molecules per vesicle, and it has been calculated that a single motor-nerve terminal contains 300,000 or more vesicles. In addition, an uncertain but possibly significant amount of ACh is present in the extravesicular cytoplasm. Recording the electrical events associated with the opening of single channels at the motor end-plate during continuous application of ACh has permitted estimation of the potential change induced by a single molecule of ACh (3 x 107 V); from such calculations, it is evident that even the lower estimate of the ACh content per vesicle (1000 molecules) is sufficient to account for the magnitude of the mepps (Katz and Miledi, 1972). The release of ACh and other neurotransmitters by exocytosis through the prejunctional membrane is inhibited by botulinum and tetanus toxins from Clostridium. Some of the most potent toxins known are produced by these spore-forming anaerobic bacteria (Shapiro et al., 1998). The Clostridium toxins, consisting of disulfide-linked heavy and light chains, bind to an as-yet-unidentified receptor on the membrane of the cholinergic nerve terminal. Through endocytosis, they are transported into the cytosol. The light chain is a Zn2+-dependent protease that becomes activated and hydrolyzes components of the core or SNARE complex involved in exocytosis. The various serotypes of botulinum toxin proteolyse selective proteins in the plasma membrane (syntaxin and SNAP-25) and the synaptic vesicle (synaptobrevin). Therapeutic uses of botulinum toxin are described in Chapters 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia and 66: Ocular Pharmacology. By contrast, tetanus toxin primarily has a central action, since it is
transported in retrograde fashion up the motor neuron to its soma in the
spinal cord. From there, the toxin migrates to inhibitory neurons that
synapse with the motor neuron and blocks exocytosis in the inhibitory neuron.
The block of release of inhibitory transmitter gives rise to tetanus or
spastic paralysis. The toxin from the venom of black widow spiders ( Characteristics of Cholinergic Transmission at Various Sites From the comparisons noted above, it is obvious that there are marked differences among various sites of cholinergic transmission with respect to architecture and fine structure, the distributions of AChE and receptors, and the temporal factors involved in normal functioning. For example, in skeletal muscle the junctional sites occupy a small, discrete portion of the surface of the individual fibers and are relatively isolated from those of adjacent fibers; in the superior cervical ganglion, approximately 100,000 ganglion cells are packed within a volume of a few cubic millimeters, and both the presynaptic and postsynaptic neuronal processes form complex networks. Skeletal Muscle Stimulation of a motor nerve results in the release of ACh from perfused muscle; close intraarterial injection of ACh produces muscular contraction similar to that elicited by stimulation of the motor nerve. The amount of ACh (1017mol) required to elicit an EPP following its microiontophoretic application to the motor end-plate of a rat diaphragm muscle fiber is equivalent to that recovered from each fiber following stimulation of the phrenic nerve (Krnjević and Mitchell, 1961). The combination of ACh with nicotinic acetylcholine receptors at the external surface of the postjunctional membrane induces an immediate, marked increase in permeability to cations. Upon activation of the receptor by ACh, its intrinsic channel opens for about 1 millisecond; during this interval about 50,000 Na+ ions traverse the channel (Katz and Miledi, 1972). The channel opening process is the basis for the localized depolarizing EPP within the end-plate, which triggers the muscle action potential. The latter, in turn, leads to contraction. Further details concerning these events and their modification by neuromuscular blocking agents are presented in Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia. Following section and degeneration of the motor nerve to skeletal muscle or of the postganglionic fibers to autonomic effectors, there is a marked reduction in the threshold doses of the transmitters and of certain other drugs required to elicit a response, i.e., denervation supersensitivity has occurred. In skeletal muscle, this change is accompanied by a spread of the receptor molecules from the end-plate region to the adjacent portions of the sarcoplasmic membrane, which eventually involves the entire muscle surface. Embryonic muscle also exhibits this uniform sensitivity to ACh prior to innervation. Hence, innervation represses the expression of the receptor gene by the nuclei that lie in extrajunctional regions of the muscle fiber and directs the subsynaptic nuclei to the expression of the structural and functional proteins of the synapse (Sanes and Lichtman, 1999). Autonomic Effectors Stimulation or inhibition of autonomic effector cells occurs upon activation of muscarinic acetylcholine receptors (see below). In this case the effector is coupled to the receptor by a G protein (see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect). In contrast to skeletal muscle and neurons, smooth muscle and the cardiac conduction system (SA node, atrium, AV node, and the His-Purkinje system) normally exhibit intrinsic activity, both electrical and mechanical, that is modulated but not initiated by nerve impulses. In the basal condition, unitary smooth muscle exhibits waves of depolarization and/or spikes that are propagated from cell to cell at rates considerably slower than the AP of axons or skeletal muscle. The spikes apparently are initiated by rhythmic fluctuations in the membrane resting potential. In intestinal smooth muscle, the site of the pacemaker activity continually shifts, but in the heart, spontaneous depolarizations normally arise from the SA node; however, when activity of the SA node is repressed or under pathological conditions, they can arise from any part of the conduction system (see Chapter 35: Antiarrhythmic Drugs). Application of ACh (0.1 to 1 In the cardiac conduction system, particularly in the SA and the AV nodes, stimulation of the cholinergic innervation or the direct application of ACh causes inhibition, associated with hyperpolarization of the membrane and a marked decrease in the rate of depolarization. These effects are due, at least in part, to a selective increase in permeability to K+ (Hille, 1992). Autonomic Ganglia The primary pathway of cholinergic transmission in autonomic ganglia is similar to that at the neuromuscular junction of skeletal muscle. Ganglion cells can be discharged by injecting very small amounts of ACh into the ganglion. The initial depolarization is the result of activation of nicotinic ACh receptors, which are ligand-gated cation channels with properties similar to those found at the neuromuscular junction. Several secondary transmitters or modulators either enhance or diminish the sensitivity of the postganglionic cell to ACh. This sensitivity appears to be related to the membrane potential of the postsynaptic nerve cell body or its dendritic branches. Ganglionic transmission is discussed in more detail in Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia. Actions of Acetylcholine at Prejunctional Sites Considerable attention has been focused on the possible involvement of prejunctional cholinoceptive sites in both cholinergic and noncholinergic transmission and in the actions of various drugs. Although cholinergic innervation of blood vessels is limited, prejunctional muscarinic receptors appear to be present on sympathetic vasoconstrictor nerves (Steinsland et al., 1973). The physiological role of these receptors is not clear, but their activation causes inhibition of neurally mediated release of norepinephrine (see Chapter 7: Muscarinic Receptor Agonists and Antagonists). Because ACh is rapidly hydrolyzed by tissue-localized and circulating esterases, it is unlikely that it plays a role as a circulating hormone analogous to that of epinephrine. Dilation of blood vessels in response to administered choline esters involves several sites of action, including prejunctional inhibitory synapses on sympathetic fibers and inhibitory cholinergic receptors in the vasculature that are not innervated. The vasodilator effect of ACh on isolated blood vessels requires an intact endothelium. Activation of muscarinic receptors results in the liberation of a vasodilator substance (endothelium-derived relaxing factor or nitric oxide) that diffuses from the endothelium to the adjoining smooth muscle and causes relaxation (see below and Chapter 7: Muscarinic Receptor Agonists and Antagonists; see also Furchgott, 1999). Cholinergic Receptors and Signal Transduction Sir Henry Dale noted that the various esters of choline elicited responses that were similar to those of either nicotine or muscarine, depending on the pharmacological preparation (Dale, 1914). A similarity in response also was noted between muscarine and nerve stimulation in those organs innervated by the craniosacral divisions of the autonomic nervous system. Thus, Dale suggested that ACh or another ester of choline was a neurotransmitter in the autonomic nervous system; he also stated that the compound had dual actions, which he termed a nicotine action (nicotinic) and a muscarine action (muscarinic). The capacities of tubocurarine and atropine to block nicotinic and muscarinic effects of ACh, respectively, provided further support for the proposal of two distinct types of cholinergic receptors. Although Dale had access only to crude plant alkaloids of then-unknown structure from Amanita muscaria and Nicotiana tabacum, this classification remains as the primary subdivision of cholinergic receptors. Its utility has survived the discovery of several distinct subtypes of nicotinic and muscarinic cholinergic receptors. Although ACh and certain other compounds stimulate both muscarinic and nicotinic receptors, several other agonists and antagonists are selective for one of the two major types of receptor. ACh itself is a flexible molecule, and indirect evidence suggests that the conformations of the neurotransmitter are distinct when it is bound to nicotinic or muscarinic receptors. Nicotinic receptors are ligand-gated ion channels, and their activation always causes a rapid (millisecond) increase in cellular permeability to Na+ and Ca2+, depolarization, and excitation. By contrast, muscarinic receptors belong to the class of G proteincoupled receptors. Responses to muscarinic agonists are slower; they may be either excitatory or inhibitory, and they are not necessarily linked to changes in ion permeability. The primary structures of various species of nicotinic receptors (Numa et al., 1983; Changeux and Edelstein, 1998) and muscarinic receptors (Bonner, 1989; Caulfield and Birdsall, 1998) have been deduced from the sequences of their respective genes. That these two types of receptor belong to distinct families of proteins is not surprising, retrospectively, in view of their distinct differences in chemical specificity and function. The nicotinic receptors exist as pentameric arrangements of one to
four distinct subunits that are homologous in sequence; the individual subunits
are arranged to surround an internal channel. One of the subunits, designated
Subtypes of Nicotinic Receptors Based on the distinct actions of certain agonists and antagonists that
interact with nicotinic receptors from skeletal muscle and ganglia, it long
has been evident that not all nicotinic receptors are identical.
Heterogeneity of this type of receptor was further revealed by molecular cloning.
For example, the muscle nicotinic receptor contains four distinct subunits in
a pentameric complex ( Subtypes of Muscarinic Receptors Five subtypes of muscarinic ACh receptors have been detected by molecular cloning. Similar to the different forms of nicotinic receptors, these variants have distinct anatomical localizations and chemical specificities. The muscarinic receptors all act through G-protein signaling systems (see discussion below and Table 62). Of the large number of muscarinic antagonists studied over many decades, only pirenzepine, found in the 1970s, showed the unique property of blocking gastric acid secretion at concentrations that did not affect several other responses to muscarinic agonists. These observations and subsequent study of other agonists and antagonists, followed by rapid advances in the cloning of cDNAs that encode muscarinic receptors, led to the identification of five subtypes of muscarinic receptors. They have been designated as M1 through M5 based on pharmacological specificity (Bonner, 1989; see also Chapter 7: Muscarinic Receptor Agonists and Antagonists). M1 receptors are found in ganglia and in some secretory glands; M2 receptors predominate in the myocardium and also appear to be found in smooth muscle; and M3 and M4 receptors are located in smooth muscle and secretory glands. All five subtypes are found in the CNS. Various tissues may contain several subtypes of muscarinic receptors; parasympathetic ganglia in the tissue also contain muscarinic receptors. The basic functions of muscarinic receptors are mediated by interactions with members of the family of G proteins and thus by G proteininduced changes in the functions of distinct membrane-bound effector molecules. The M1, M3, and M5 subtypes activate a G protein, termed Gq/11, that is responsible for stimulation of phospholipase C activity; the immediate result is hydrolysis of phosphatidylinositol polyphosphates (which are components of the plasma membrane) to form inositol polyphosphates. Some of the inositol phosphate isomers (chiefly inositol-1,4,5-trisphosphate) cause release of intracellular Ca2+ from stores in the endoplasmic reticulum. Thus, these receptors mediate such Ca2+-dependent phenomena as contraction of smooth muscle and secretion (see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect; see also Berridge, 1993). The second product of the phospholipase C reaction, diacylglycerol, activates protein kinase C (in conjunction with Ca2+). This arm of the pathway plays a role in modulation of function and in the later phases of the functional response (Dempsey et al., 2000). A second pathway for mediation of responses to muscarinic agonists is evoked by activation of M2 and M4 receptors. These receptors interact with a distinct group of G proteins (in particular those termed Gi and Go) with resultant inhibition of adenylyl cyclase, activation of receptor-operated K+ channels (in the heart, for example), and suppression of the activity of voltage-gated Ca2+ channels in certain cell types. The functional consequences of these effects are most clear in the myocardium, where inhibition of adenylyl cyclase and activation of K+ conductances can account for both the negative chronotropic and inotropic effects of ACh. Other cellular events such as the release of arachidonic acid and the activation of guanylyl cyclase also can result from activation of muscarinic receptors; typically these responses are secondary to the production of other mediators. Adrenergic Transmission Under this general heading are included norepinephrine, the transmitter of most sympathetic postganglionic fibers and of certain tracts in the CNS, and dopamine, the predominant transmitter of the mammalian extrapyramidal system and of several mesocortical and mesolimbic neuronal pathways, as well as epinephrine, the major hormone of the adrenal medulla. A tremendous amount of information about catecholamines and related compounds has accumulated in recent years, partly because of the importance of interactions between the endogenous catecholamines and many of the drugs used in the treatment of hypertension, mental disorders, and a variety of other conditions. The details of these interactions and of the pharmacology of the sympathomimetic amines themselves will be found in subsequent chapters. The basic physiological, biochemical, and pharmacological features are presented here. Synthesis, Storage, and Release of Catecholamines Synthesis The synthesis of epinephrine from tyrosine, by the steps shown in Figure
63, was proposed by Blaschko in 1939. The enzymes involved have been
identified, cloned, and characterized (Nagatsu, 1991). It is important to
note that these enzymes are not completely specific; consequently, other
endogenous substances as well as certain drugs are similarly acted upon at
the various steps. For example, 5-hydroxytryptamine (5-HT, serotonin) can be
produced by aromatic L-amino acid decarboxylase (or dopa decarboxylase) from 5-hydroxy-L-tryptophan. Dopa decarboxylase
also converts dopa into dopamine, and methyldopa is converted to
The h Current knowledge concerning the cellular sites and mechanisms of
synthesis, storage, and release of catecholamines has been derived from
studies of both adrenergically innervated organs and of adrenal medullary
tissue. Nearly all the norepinephrine content of the former is confined to the
postganglionic sympathetic fibers; it disappears within a few days after
section of the nerves. In the adrenal medulla, catecholamines are stored in
chromaffin granules (Winkler, 1997; Aunis, 1998). These vesicles contain
extremely high concentrations of catecholamines (approximately 21% dry
weight), ascorbic acid, and ATP, as well as specific proteins such as
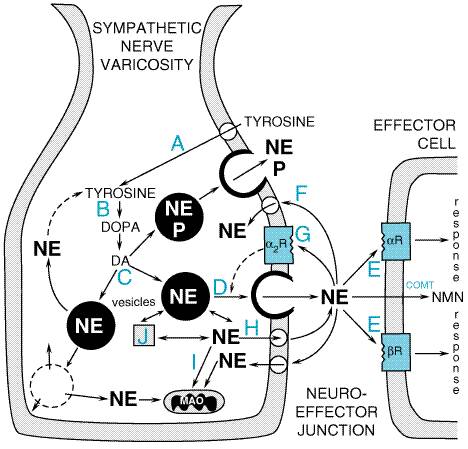
chromogranins, the enzyme dopamine The main features of the mechanisms of synthesis, storage, and release of catecholamines and their modifications by drugs are summarized in Figure 64. In the case of adrenergic neurons, the enzymes that participate in the formation of norepinephrine are synthesized in the cell bodies of the neurons and are then transported along the axons to their terminals. In the course of synthesis (see Figure 63), the hydroxylation of tyrosine to dopa and the decarboxylation of dopa to dopamine take place in the cytoplasm. About half the dopamine formed in the cytoplasm then is actively transported into the DBH-containing storage vesicles, where it is converted to norepinephrine; the remainder, which escaped capture by the vesicles, is deaminated to 3,4-dihydroxyphenylacetic acid (DOPAC) and subsequently O-methylated to homovanillic acid (HVA). The adrenal medulla has two distinct catecholamine-containing cell types: those with norepinephrine and those with primarily epinephrine. The latter cell population contains the enzyme phenylethanolamine-N-methyltransferase. In these cells, the norepinephrine formed in the granules leaves these structures, presumably by diffusion, and is methylated in the cytoplasm to epinephrine. Epinephrine then reenters the chromaffin granules, where it is stored until released. In adults, epinephrine accounts for approximately 80% of the catecholamines of the adrenal medulla, with norepinephrine making up most of the remainder (von Euler, 1972).
A major factor that controls the rate of synthesis of epinephrine, and hence the size of the store available for release from the adrenal medulla, is the level of glucocorticoids secreted by the adrenal cortex. The latter hormones are carried in high concentration, by the intraadrenal portal vascular system, directly to the adrenal medullary chromaffin cells, where they induce the synthesis of phenylethanolamine-N-methyltransferase (see Figure 63). The activities of both tyrosine hydroxylase and DBH also are increased in the adrenal medulla when the secretion of glucocorticoids is stimulated (Carroll et al., 1991; Viskupic et al., 1994). Thus, any stress that persists sufficiently to evoke an enhanced secretion of corticotropin mobilizes the appropriate hormones of both the adrenal cortex (predominantly cortisol) and medulla (epinephrine). This remarkable relationship is present only in certain mammals, including human beings, for which the adrenal chromaffin cells are enveloped entirely by steroid-secreting cortical cells. In the dogfish, for example, where the chromaffin cells and steroid-secreting cells are located in independent, noncontiguous glands, epinephrine is not formed. Nonetheless, there is evidence indicating that phenylethanolamine-N-methyltransferase is expressed in mammalian tissues such as brain, heart, and lung, leading to extra-adrenal epinephrine synthesis (Kennedy and Ziegler, 1991; Kennedy et al., 1993). In addition to its de novo synthesis, outlined above, there is a second major mechanism for replenishment of the norepinephrine stored in the terminal portions of the adrenergic fibersnamely, recapture by active transport of norepinephrine previously released to the extracellular fluid. This process is responsible for the termination of the effects of adrenergic impulses in most organs. In blood vessels and in tissues with wide synaptic gaps, recapture of released norepinephrine is less important. At such sites, a relatively large fraction of the released neurotransmitter is inactivated by a combination of extraneuronal uptake (see below) and enzymatic breakdown and diffusion. To effect the reuptake of norepinephrine into adrenergic nerve terminals and to maintain the concentration gradient of norepinephrine within the vesicles, at least two distinct carrier-mediated transport systems are involved: one across the axoplasmic membrane from the extracellular fluid to the cytoplasm and the other from the cytoplasm into the storage vesicles. Storage of Catecholamines Catecholamines are stored in vesicles to ensure their regulated release; this storage decreases intraneuronal metabolism of these transmitters as well as their leakage outside the cell. The amine transporter has been extensively characterized (Schuldiner, 1994). Uptake of catecholamine and ATP into isolated chromaffin granules appears to be driven by pH and potential gradients that are established by an ATP-dependent proton translocase. For every molecule of amine taken up, two H+ ions are extruded (Brownstein and Hoffman, 1994). Monoamine transporters are relatively promiscuous, capable of transporting dopamine, norepinephrine, epinephrine, and serotonin, for example. Also, meta-iodobenzylguanidine, used clinically to image chromaffin-cell tumors, is a substrate for this transport system (Schuldiner, 1994). Reserpine is a drug that inhibits monoamine transport into these vesicles which ultimately leads to depletion of catecholamine from sympathetic nerve endings and in the brain. Several vesicular transport cDNAs have been identified with molecular cloning techniques; these cDNAs reveal open reading frames predictive of proteins with 12 putative transmembrane domains with structural homology to other transport proteins such as bacterial drug-resistance transporters (Schuldiner, 1994). Regulation of the expression of these various transporters may be important in the regulation of synaptic transmission (Varoqui and Erickson, 1997). When catecholamines such as norepinephrine are injected into the blood
of experimental animals, they are rapidly accumulated in tissues with
extensive sympathetic innervation, such as heart and spleen; labeled
catecholamines are concentrated in sympathetic nerve endings, and tissue
uptake disappears after denervation (reviewed in Brownstein and Hoffman, 1994).
This and other evidence suggested the presence of transporters on the plasma
membrane of sympathetic neurons that could take up catecholamines. The amine
transport system across the axoplasmic membranes is Na+-dependent
and is blocked selectively by a number of drugs, including cocaine and the
tricyclic antidepressants, such as imipramine. This transporter has a high
affinity for norepinephrine and a somewhat lower affinity for epinephrine;
the synthetic Certain sympathomimetic drugs (e.g., ephedrine, tyramine) produce some of their effects indirectly, chiefly by displacing norepinephrine from the nerve-ending binding sites to the extracellular fluid, where the released endogenous transmitter then acts at receptor sites of the effector cells. The mechanisms by which indirect-acting sympathomimetic amines release norepinephrine from nerve endings are complex. All such agents are substrates for uptake-1. As a result of their transport across the neuronal membrane and release into the axoplasm, they make carrier available at the inner surface of the membrane for the outward transport of norepinephrine ('facilitated exchange diffusion'). In addition, these amines are able to mobilize norepinephrine stored in the vesicles by competing for the vesicular uptake process. Reserpine, which depletes vesicular stores of norepinephrine, also inhibits this uptake mechanism, but, in contrast with the indirect-acting sympathomimetic amines, it enters the adrenergic nerve ending by passive diffusion across the axonal membrane (Bnisch and Trendelenburg, 1988). These actions of indirect-acting sympathomimetic amines are associated
with the phenomenon of tachyphylaxis. For example, repeated
administration of tyramine results in rapidly decreasing effectiveness,
whereas repeated administration of norepinephrine does not reduce
effectiveness and, in fact, reverses the tachyphylaxis to tyramine. Although
these phenomena have not been explained fully, several hypotheses have been
proposed. One possible explanation of tachyphylaxis to tyramine and similarly
acting sympathomimetic agents is that the pool of neurotransmitter available
for displacement by these drugs is small relative to the total amount stored
in the sympathetic nerve ending. This pool is presumed to reside in close
proximity to the plasma membrane, and the norepinephrine of such vesicles may
be replaced by the less potent amine following repeated administration of the
latter substance. In any case, neurotransmitter release by displacement is
not associated with the release of dopamine There also is an extraneuronal amine transport system, termed uptake-2, which exhibits a low affinity for norepinephrine, a somewhat higher affinity for epinephrine, and a still higher affinity for isoproterenol. This uptake process is ubiquitous and is present in glial, hepatic, myocardial, and other cells. Uptake-2 is not inhibited by imipramine or cocaine. It probably is of relatively little physiological importance unless the neuronal uptake mechanism is blocked (Iversen, 1975; Trendelenburg, 1980). It may be of greater importance in the disposition of circulating catecholamines than in the removal of amines that have been released from adrenergic nerve terminals. Release of Catecholamines The full sequence of steps by which the nerve impulse effects the
release of norepinephrine from adrenergic fibers is not known. In the adrenal
medulla, the triggering event is the liberation of ACh by the preganglionic fibers
and its interaction with nicotinic receptors on chromaffin cells to produce a
localized depolarization; a succeeding step is the entrance of Ca2+
into these cells, which results in the extrusion by exocytosis of the
granular contents, including epinephrine, ATP, some neuroactive peptides or
their precursors, chromogranins, and DBH. Influx of Ca2+ likewise
plays an essential role in coupling the nerve impulse, membrane
depolarization, and opening of voltage-gated Ca2+ channels with
the release of norepinephrine at adrenergic nerve terminals. Blockade of
N-type Ca2+ channels leads to hypotension, likely due to
inhibition of norepinephrine release (Bowersox et al., 1992). Ca2+-triggered
secretion involves interaction of highly conserved molecular scaffolding
proteins leading to docking of granules at the plasma membrane, ultimately
leading to secretion ( Adrenergic fibers can sustain the output of norepinephrine during
prolonged periods of stimulation without exhausting their reserve supply,
provided synthesis and uptake of the transmitter are unimpaired. To meet
increased needs for norepinephrine, acute regulatory mechanisms come into
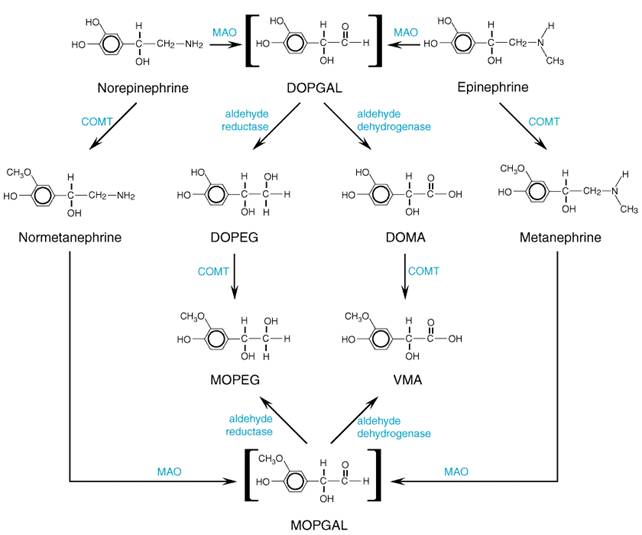
play that involve activation of tyrosine hydroxylase and dopamine Termination of the Actions of Catecholamines The actions of norepinephrine and epinephrine are terminated by (1) reuptake into nerve terminals; (2) dilution by diffusion out of the junctional cleft and uptake at extraneuronal sites; and (3) metabolic transformation. Two enzymes are important in the initial steps of metabolic transformation of catecholaminesmonoamine oxidase (MAO) and catechol-O-methyltransferase (COMT; see Axelrod, 1966; Kopin, 1972). In addition, catecholamines are metabolized by sulfotransferases (Dooley, 1998). However, it is evident that a powerful degradative enzymatic pathway, such as that provided by AChE, is absent from the adrenergic nervous system. The importance of neuronal reuptake of catecholamines is shown by observations that inhibitors of this process (e.g., cocaine, imipramine) potentiate the effects of the neurotransmitter; inhibitors of MAO and COMT have relatively little effect. However, transmitter that is released within the nerve terminal is metabolized by MAO. COMT, particularly in the liver, plays a major role in the metabolism of endogenous circulating and administered catecholamines. Both MAO and COMT are widely distributed throughout the body, including the brain; the highest concentrations of each are in the liver and the kidney. However, little or no COMT is found in adrenergic neurons. There are distinct differences in the cytological locations of the two enzymes; whereas MAO is associated chiefly with the outer surface of mitochondria, including those within the terminals of adrenergic fibers, COMT is located largely in the cytoplasm. These factors are of importance both in determining the primary metabolic pathways followed by catecholamines in various circumstances and in explaining the effects of certain drugs. Two different isozymes of MAO (MAO-A and MAO-B) are found in widely varying proportions in different cells in the CNS and in peripheral tissues. Selective inhibitors of these two isozymes are available (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). Irreversible antagonists of MAO-A enhance the bioavailability of tyramine contained in many foods; tyramine-induced norepinephrine release from sympathetic neurons may lead to markedly increased blood pressure. Selective MAO-B inhibitors (e.g., selegiline) or reversible MAO-A-selective inhibitors (moclobemide) are less likely to cause this potential interaction (Volz and Geiter, 1998; Wouters, 1998). MAO inhibitors are useful in the treatment of Parkinson's disease and mental depression (see Chapters 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders and 22: Treatment of Central Nervous System Degenerative Disorders). Most of the epinephrine and norepinephrine that enter the circulationfrom the adrenal medulla or following administration or that is released by exocytosis from adrenergic fibersis methylated by COMT to metanephrine or normetanephrine, respectively (Figure 65). Norepinephrine that is released intraneuronally by drugs such as reserpine is initially deaminated by MAO to 3,4-dihydroxyphenylglycolaldehyde (DOPGAL; see Figure 65). The aldehyde is reduced by aldehyde reductase to the glycol, 3,4-dihydroxyphenylethylene glycol (DOPEG), or is oxidized by aldehyde dehydrogenase to form 3,4-dihydroxymandelic acid (DOMA). 3-Methoxy-4-hydroxymandelic acid [generally but incorrectly called vanillylmandelic acid (VMA)] is the major metabolite of catecholamines excreted in the urine. The corresponding product of the metabolic degradation of dopamine, which contains no hydroxyl group in the side chain, is homovanillic acid (HVA). Other metabolic reactions are described in Figure 65. Measurement of the concentrations of catecholamines and their metabolites in blood and urine is useful in the diagnosis of pheochromocytoma, a catecholamine-secreting tumor of the adrenal medulla.
Inhibitors of MAO (e.g., pargyline, nialamide) can cause an increase in the concentration of norepinephrine, dopamine, and 5-HT in the brain and other tissues accompanied by a variety of pharmacological effects. No striking pharmacological action in the periphery can be attributed to the inhibition of COMT. However, a COMT inhibitor, entacapone, has been found to be efficacious in the therapy of Parkinson's disease (Chong and Mersfelder, 2000; see also Chapter 22: Treatment of Central Nervous System Degenerative Disorders). Classification of Adrenergic Receptors Crucial to understanding the remarkably diverse effects of the catecholamines and related sympathomimetic agents is an understanding of the classification and properties of the different types of adrenergic receptors (or adrenoceptors). Elucidation of the characteristics of these receptors and the biochemical and physiological pathways they regulate has increased our understanding of the seemingly contradictory and variable effects of catecholamines on various organ systems. Although structurally related (see below), different adrenergic receptors regulate distinct physiological processes by controlling the synthesis or release of a variety of second messengers (see Tables 63 and 64). Ahlquist (1948) first proposed the existence of more than one
adrenergic receptor; he based his hypothesis on a study of the abilities of epinephrine,
norepinephrine, and other related agonists to regulate various physiological
processes. It was known that these drugs could cause either contraction or
relaxation of smooth muscle, depending on the site, the dose, and the agent
chosen. For example, norepinephrine was known to have potent excitatory
effects on smooth muscle and correspondingly low activity as an inhibitor; isoproterenol
displayed the opposite pattern of activity. Epinephrine could both excite and
inhibit smooth muscle. Thus, Ahlquist proposed the designations
The heterogeneity of Cloning revealed additional heterogeneity of both Molecular Basis of Adrenergic Receptor Function The responses that follow activation of all types of adrenergic receptors appear to result from G proteinmediated effects on the generation of second messengers and on the activity of ion channels. As discussed in Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect, these systems involve three interacting proteinsthe receptor, the coupling G protein, and effector enzymes or ion channels. The pathways overlap broadly with those discussed for muscarinic receptors and are summarized in Table 64. Structure of Adrenergic Receptors The adrenergic receptors constitute a family of closely related proteins. They also are related both structurally and functionally to receptors for a wide variety of other hormones and neurotransmitters that are coupled to G proteins (Lefkowitz, 2000). This wider family of receptors includes the muscarinic acetylcholine receptors and even the visual 'photon receptor,' rhodopsin (see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect). Ligand binding, site-directed labeling, and mutagenesis have revealed that the conserved membrane-spanning regions are crucially involved in the binding of ligands (Strader et al., 1994; Hutchins, 1994). These regions appear to create a ligand-binding pocket analogous to that formed by the membrane-spanning regions of rhodopsin to accommodate the covalently attached chromophore, retinal, with molecular models placing catecholamines either horizontally (Strader et al., 1994) or perpendicularly (Hutchins, 1994) in the bilayer. The crystal structure of mammalian rhodopsin has been determined and confirms a number of predictions about the structure of G proteincoupled receptors (Palczewski et al., 2000).
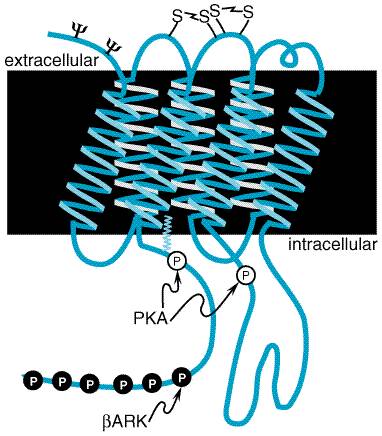
The three All The cyclic AMPdependent protein kinase (protein kinase A) generally
is considered the major intracellular receptor for cyclic AMP. It exists as a
tetramer (R2C2), consisting of two regulatory (R) and
two catalytic (C) subunits. Binding of cyclic AMP causes dissociation of the
regulatory subunits, as a result of a 10,000- to 100,000-fold decrease in
affinity of R for C, with resultant activation of the catalytic subunits (Francis
and Corbin, 1994; Smith et al., 1999). Phosphorylation of various
cellular proteins then causes responses that are characteristic of those
produced by A well-defined example of these mechanisms is the activation of hepatic glycogen phosphorylase, the enzyme that promotes the rate-limiting step in glycogenolysis, the conversion of glycogen to glucose-1-phosphate. This activation is itself the result of a cascade of phosphorylation reactions. Protein kinase A catalyzes the phosphorylation of phosphorylase kinase, thereby activating it; phosphorylase kinase then phosphorylates and activates phosphorylase. This sequence of successive phosphorylations permits considerable amplification of the initial signal. Thus, only a few receptors need to be stimulated to activate a large number of phosphorylase molecules in a very brief period of time. Concurrent with the activation of hepatic glycogen phosphorylase, protein kinase A also catalyzes the phosphorylation and inactivation of another enzyme, glycogen synthase, which catalyzes the transfer of glycosyl units from UDP-glucose to glycogen. Phosphorylation decreases the net rate of synthesis of glycogen from glucose. The dual effects of cyclic AMP to enhance conversion of glycogen to glucose and to decrease the synthesis of glycogen from glucose summate to increase the output of glucose from the liver. Similar types of reactions result in the activation of triglyceride lipase in adipose tissue, with resultant increased release of free fatty acids. The lipase is activated when it is phosphorylated by protein kinase A. Catecholamines provide an increased supply of substrate for oxidative metabolism by this mechanism. In the heart, stimulation of
The deduced amino acid sequences from the six
As shown in Table 64,
Stimulation of
In most smooth muscles, the increased concentrations of intracellular
Ca2+ ultimately cause contraction as a result of activation of Ca2+-sensitive
protein kinases such as the calmodulin-dependent myosin light chain kinase;
phosphorylation of the light chain of myosin is associated with the
development of tension (Stull et al., 1990). In contrast, the
increased concentrations of intracellular Ca2+ that result from
stimulation of As with Localization of Adrenergic Receptors Presynaptically located In contrast, The cellular distributions of the three Refractoriness to Catecholamines Exposure of catecholamine-sensitive cells and tissues to adrenergic
agonists causes a progressive diminution in their capacity to respond to such
agents. This phenomenon is variously termed refractoriness,
desensitization, or tachyphylaxis, and it significantly limits the
therapeutic efficacy and duration of action of catecholamines and other
agents (see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and
the Relationship Between Drug Concentration and Effect). Although
descriptions of such adaptive changes are common, mechanisms are incompletely
understood. They have been studied most extensively in cells that synthesize
cyclic AMP in response to There is evidence for multiple points of regulation of responsiveness,
including the receptors, G proteins, adenylyl cyclase, and cyclic nucleotide
phosphodiesterase. The pattern of refractoriness varies according to the
extent to which these different components are modified. In some cases,
especially when the receptors themselves are altered, desensitization may be
limited to the actions of One of the most important mechanisms for rapidly regulating Mechanisms for Heterologous Desensitization One protein kinase that phosphorylates Gs-coupled receptors
is protein kinase A, which is stimulated by
Mechanisms for Homologous Desensitization A receptor-directed protein kinase, termed the GRK3 also contains a Unlike the situation with protein kinase A-mediated receptor
phosphorylation, covalent modification of the G proteincoupled receptor by
GRKs alone is not sufficient to fully desensitize receptor function. Rather,
a second reaction must occur in which an 'arresting' protein binds
to the phosphorylated receptor and presumably sterically inhibits its
functional coupling to Gs. This protein, called Agonists also promote a rapid (minutes) and reversible sequestration
(internalization) of their receptors and a slower (hours)
'down-regulation' of the receptors in which the actual number of
receptors in the cell declines. The function of receptor sequestration is not
fully understood. Interestingly, there is evidence suggesting that receptor
internalization is important for some (Daaka et al., 1998) but not all
G proteincoupled receptor-mediated activation of MAP kinase (Schramm and
Limbird, 1999; Pierce et al., 2000). Quantitatively, sequestration may
not contribute significantly to the mechanisms underlying desensitization,
particularly because there is a high degree of amplification between There is evidence that sequestration, internalization, and
down-regulation may occur for |
Relationship between the Nervous and the Endocrine Systems
|
The concept that 'humours' are secreted at certain sites to act elsewhere in the body can be traced back to Aristotle. In modern terms, the theory of neurohumoral transmission by its very designation implies at least a superficial resemblance between the nervous and the endocrine systems. Yet it should now be clear that the similarities extend considerably deeper, particularly with respect to the autonomic nervous system. In the regulation of homeostasis, the autonomic nervous system is responsible for rapid adjustments to changes in the total environment, which it effects at both its ganglionic synapses and postganglionic terminals by the liberation of chemical agents that act transiently at their immediate sites of release. The endocrine system, in contrast, regulates slower, more generalized adaptations by releasing hormones into the systemic circulation to act at distant, widespread sites over periods of minutes to hours or days. Both systems have major central representations in the hypothalamus, where they are integrated with each other and with subcortical, cortical, and spinal influences. The neurohumoral theory thus may be said to provide a unitary concept of the functioning of the nervous and endocrine systems, in which the differences largely relate to the distances over which the released mediators travel. |
Pharmacological Considerations
|
The foregoing sections contain numerous references to the actions of drugs considered primarily as tools for the dissection and elucidation of physiological mechanisms. This section presents a classification of drugs that act on the peripheral nervous system and its effector organs at some stage of neurotransmission. In the immediately succeeding chapters, as well as elsewhere in the text, the systematic pharmacology of the important members of each of these classes is described. Each step involved in neurotransmission (see Figure 62) represents a potential point of therapeutic intervention. This is depicted in the diagram of the adrenergic terminal and its postjunctional site (Figure 64). Drugs that affect processes involved in each step of transmission at both cholinergic and adrenergic junctions are summarized in Table 66, which lists representative agents that act via the mechanisms described below. Interference with the Synthesis or Release of the Transmitter Cholinergic Hemicholinium (HC-3), a synthetic compound, blocks the transport system by which choline accumulates in the terminals of cholinergic fibers, thus limiting the synthesis of the ACh store available for release (Birks and MacIntosh, 1957). Vesamicol blocks the transport of ACh into its storage vesicles, preventing its release. The site on the presynaptic nerve terminal for block of ACh release by botulinum toxin was discussed previously; death usually results from respiratory paralysis, unless patients with respiratory failure receive artificial ventilation. Injected locally, botulinum toxin is used in the treatment of muscle dystonias, palsy (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia), certain ophthalmological conditions associated with spasms of ocular muscles (see Chapter 66: Ocular Pharmacology), or for treatment of anal fissures. Adrenergic
Promotion of the Release of the Transmitter Cholinergic The ability of cholinergic agents to promote the release of ACh is limited, presumably because ACh and other cholinomimetic agents are quaternary ammonium compounds and do not readily cross the axonal membrane into the nerve ending. The latrotoxins from black widow spider venom and stonefish are known to promote neuroexocytosis by binding to receptors on the neuronal membrane. Adrenergic Several drugs that promote the release of the adrenergic mediator lready have been discussed. On the basis of the rate and the duration of the drug-induced release of norepinephrine from adrenergic terminals, one of two opposing effects can predominate. Thus, tyramine, ephedrine, amphetamine, and related drugs cause a relatively rapid, brief liberation of the transmitter and produce a sympathomimetic effect. On the other hand, reserpine, by blocking vesicular uptake of amines, produces a slow, prolonged depletion of the adrenergic transmitter from adrenergic storage vesicles, where it is largely metabolized by intraneuronal MAO. The resultant depletion of transmitter produces the equivalent of adrenergic blockade. Reserpine also causes the depletion of serotonin, dopamine, and possibly other, unidentified amines from central and peripheral sites, and many of its major effects may be a consequence of the depletion of transmitters other than norepinephrine. A syndrome caused by congenital dopamine- Agonist and Antagonist Actions at Receptors Cholinergic The nicotinic receptors of autonomic ganglia and skeletal muscle are not identical; they respond differently to certain stimulating and blocking agents and their pentameric structures contain different combinations of homologous subunits (see Table 62). Dimethylphenylpiperazinium (DMPP) and phenyltrimethylammonium (PTMA) show some selectivity for stimulation of autonomic ganglion cells and end-plates of skeletal muscle, respectively. Trimethaphan and hexamethonium are relatively selective competitive and noncompetitive ganglionic blocking agents. Although tubocurarine effectively blocks transmission at both motor end-plates and autonomic ganglia, its action at the former site predominates. Decamethonium, a depolarizing agent, produces selective neuromuscular blockade. Transmission at autonomic ganglia and the adrenal medulla is complicated further by the presence of muscarinic receptors, in addition to the principal nicotinic receptors (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). Various toxins in snake venoms exhibit a high degree of specificity in
the cholinergic nervous system. The Muscarinic receptors, which mediate the effects of ACh at autonomic effector cells, now can be divided into five subclasses. Atropine blocks all the muscarinic responses to injected ACh and related cholinomimetic drugs, whether they are excitatory, as in the intestine, or inhibitory, as in the heart. Newer muscarinic agonists, pirenzepine for M1, tripitramine for M2, and darifenacin for M3, show selectivity as muscarinic blocking agents. Several muscarinic antagonists show sufficient selectivity in clinical setting to minimize the bothersome side effects seen with the nonselective agents at therapeutic doses (see Chapter 7: Muscarinic Receptor Agonists and Antagonists). Adrenergic A vast number of synthetic compounds that bear structural resemblance
to the naturally occurring catecholamines can interact with Interference with the Destruction of the Transmitter Cholinergic The anti-ChE agents (Chapter 8: Anticholinesterase Agents) constitute a chemically diverse group of compounds, the primary action of which is inhibition of AChE, with the consequent accumulation of endogenous ACh. At the neuromuscular junction, accumulation of ACh produces depolarization of end-plates and flaccid paralysis. At postganglionic muscarinic effector sites, the response is either excessive stimulation resulting in contraction and secretion or an inhibitory response mediated by hyperpolarization. At ganglia, depolarization and enhanced transmission are observed. Adrenergic The reuptake of norepinephrine by the adrenergic nerve terminals probably is the major mechanism for terminating its transmitter action. Interference with this process is the basis of the potentiating effect of cocaine on responses to adrenergic impulses and injected catecholamines. It also has been suggested that the antidepressant actions and some of the adverse effects of imipramine and related drugs are due to a similar action at adrenergic synapses in the CNS (see Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). Inhibitors of COMT, such as tolcapone, enhance dopamine action in the brain of patients with Parkinson's disease (see Chapter 22: Treatment of Central Nervous System Degenerative Disorders). On the other hand, MAO inhibitors, such as tranylcypromine, potentiate the effects of tyramine and may potentiate effects of neurotransmitters. |
Other Autonomic Neurotransmitters
|
Evidence has accumulated in recent years that the vast majority of neurons in both the central and peripheral nervous systems contain more than one substance with potential or demonstrated activity at relevant postjunctional sites (see Bartfai et al., 1988; Bennett, 1997; Lundberg, 1996; see also Chapter 12: Neurotransmission and the Central Nervous System). In some cases, especially in peripheral structures, it has been possible to demonstrate that two or more such substances are contained within individual nerve terminals and are released simultaneously upon nerve stimulation. Although the anatomical separation of the parasympathetic and sympathetic components of the autonomic nervous system and the actions of ACh and norepinephrine (their primary neurotransmitters) still provide the essential framework for studying autonomic function, a host of other chemical messengers such as purines, eicosanoids, nitric oxide, and peptides modulate or mediate responses that follow stimulation of the preganglionic neurons of the autonomic nervous system. An expanded view of autonomic neurotransmission has evolved to include instances where substances other than ACh or norepinephrine are released and may function as cotransmitters, neuromodulators, or even primary transmitters. Moreover, vagal fibers innervating the gastrointestinal tract synapse with excitatory and inhibitory postganglionic neurons, allowing for both intrinsic excitation and inhibition during a peristalic wave and inhibition of sphincters. The evidence for cotransmission, or for so-called nonadrenergic, noncholinergic transmission, in the autonomic nervous system usually encompasses the following considerations: (1) A portion of responses to stimulation of preganglionic or postganglionic nerves or to field stimulation of target structures persists in the presence of concentrations of muscarinic or adrenergic antagonists that completely block their respective agonists. (2) The candidate substance can be detected within nerve fibers that course through target tissues. (3) The substance can be recovered upon microdialysis or in the venous or perfusion effluent following electrical stimulation; such release often can be blocked by tetrodotoxin. (4) Effects of electrical stimulation are mimicked by the application of the substance and are inhibited in the presence of specific antagonists. When such antagonists are not available, reliance often is placed on neutralizing antibodies or selective desensitization produced by prior exposure to the substance. A more recent approach to this challenging problem is the use of 'knockout' mice which do not express the putative cotransmitter. A number of problems confound interpretation of such evidence. It is particularly difficult to establish that substances that fulfill all the listed criteria originate within the autonomic nervous system. In some instances, their origin can be traced to sensory fibers, to intrinsic neurons, or to nerves innervating blood vessels. Also, there may be marked synergism between the candidate substance and known or unknown transmitters (Lundberg, 1996). In knockout mice, compensatory mechanisms or transmitter redundancy may disguise even well-defined actions (Hkfelt et al., 2000). Finally, it should be recognized that the putative cotransmitter may have primarily a trophic function in maintaining synaptic connectivity or in expressing a particular receptor. It long has been known that ATP and ACh coexist in cholinergic vesicles (Dowdall et al., 1974) and that ATP and catecholamines both are found within storage granules in nerves and the adrenal medulla (see above). ATP is released along with the transmitters, and either it or its metabolites have a significant function in synaptic transmission in some circumstances (see below). More recently, attention has focused on the growing list of peptides that are found in the adrenal medulla, nerve fibers or ganglia of the autonomic nervous system, or in the structures that are innervated by the autonomic nervous system. This list includes the enkephalins, substance P and other tachykinins, somatostatin, gonadotropin-releasing hormone, cholecystokinin, calcitonin generelated peptide, galanin, pituitary adenylyl cyclase-activating peptide, VIP, chromogranin, and neuropeptide Y (NPY) (Darlison and Richter, 1999; Lundberg, 1996; Bennett, 1997, Hkfelt et al., 2000). Some of the orphan G proteincoupled receptors discovered in the course of genome sequencing projects may represent receptors for as yet undiscovered peptides or other cotransmitters. The evidence for widespread transmitter function in the autonomic nervous system is substantial for VIP and NPY, and further discussion is confined to these peptides. The possibility that abnormalities in function of neuropeptide cotransmitters, for example in type 2 diabetes, contribute to disease pathogenesis remains of interest (Ahren, 2000). Cotransmission in the Autonomic Nervous System Both norepinephrine and ATP elicit excitation when released from certain adrenergic nerve terminals, such as those in the vas deferens and the vasculature. The response to ATP is rapid and that to norepinephrine is slower (Sneddon and Westfall, 1984). Sympathectomy and adrenergic neurondepleting agents such as reserpine eliminate both phases of the response, consistent with storage of both substances in the same population of vesicles. In other cases, metabolism of ATP to adenosine in the extracellular space results in important modulatory effects. There also is evidence that adenosine exerts inhibitory effects on the release of transmitter, and the administration of adenosine-receptor antagonists such as theophylline results in increased concentrations of norepinephrine and other components of the storage vesicle in the circulation. The pioneering studies of Hkfelt and coworkers (Lundberg et al., 1979), which demonstrated the existence of VIP and ACh in peripheral autonomic neurons, initiated interest in the possibility of peptidergic cotransmission in the autonomic nervous system. Subsequent work has confirmed the frequent association of these two substances in autonomic fibers, including parasympathetic fibers that innervate smooth muscle and exocrine glands and cholinergic sympathetic neurons that innervate sweat glands (Hkfelt et al., 2000). The role of VIP in parasympathetic transmission has been most extensively studied in the regulation of salivary secretion. The evidence for cotransmission includes the release of VIP following stimulation of the chorda lingual nerve and the incomplete blockade by atropine of vasodilation when the frequency of stimulation is raised; the latter observation may indicate independent release of the two substances, which is consistent with histochemical evidence for storage of ACh and VIP in separate populations of vesicles. Synergism between ACh and VIP in stimulating vasodilation and secretion also has been described. VIP may be involved in parasympathetic responses in the trachea and the gastrointestinal tract; in the latter it may facilitate sphincter relaxation. The neuropeptide Y family of peptides is widely distributed in the central and peripheral nervous systems and consists of three members: NPY, pancreatic polypeptide, and peptide YY. In the CNS, NPY function is linked to food and water intake, regulation of mood, and central autonomic control. In the periphery, NPY is found in large vesicles within sympathetic nerve fibers and is involved in the maintenance of vascular tone. NPY and norepinephrine are coreleased, although low-frequency stimulation may favor norepinephrine release. NPY has a potent and prolonged vasoconstrictor action, with small blood vessels being more sensitive. Its activity appears to be synergistic with that of norepinephrine. Multiple subtypes of NPY receptors have been identified and cloned; all appear to function through G proteins (Wahlestedt and Reis, 1993). The role of NPY, especially including its regulation of leptin and regulation of appetite and body weight, offers the potential for discovery of novel drugs for the treatment of obesity (Good, 2000; Poyner et al., 2000; Halford and Blundell, 2000). Nonadrenergic, Noncholinergic Transmission by Purines The smooth muscle of many tissues that are innervated by the autonomic nervous system shows inhibitory junction potentials following stimulation by field electrodes (Bennett, 1997). Since such responses frequently are undiminished in the presence of adrenergic and muscarinic cholinergic antagonists, these observations have been taken as evidence for the existence of nonadrenergic, noncholinergic transmission in the autonomic nervous system. Burnstock (1969, 1996) and his colleagues have compiled compelling evidence for the existence of purinergic neurotransmission in the gastrointestinal tract, genitourinary tract, and certain blood vessels; ATP has fulfilled all the criteria for a neurotransmitter listed above. However, in at least some circumstances, primary sensory axons may be an important source of ATP (Burnstock, 2000). Although adenosine is generated from the released ATP by ectoenzymes, its primary function appears to be modulatory by causing feedback inhibition of release of the transmitter. Adenosine can be transported from the cell cytoplasm to activate extracellular receptors on adjacent cells. The efficient uptake of adenosine by cellular transporters and its rapid rate of metabolism to inosine or to adenine nucleotides contribute to its rapid turnover. Several inhibitors of adenosine transport and metabolism are known to influence extracellular adenosine and ATP concentrations (Sneddon et al., 1999). The purinergic receptors found on the cell surface may be divided into the adenosine (A or P1) and the ATP (P2) receptors (Fredholm et al., 1997). Each of P1 and P2 receptors has been found to have various subtypes. Methylxanthines such as caffeine and theophylline preferentially block adenosine receptors (see Chapter 28: Drugs Used in the Treatment of Asthma). At least seven subtypes of both P1 and P2 receptors have been identified in brain, peripheral tissues, and circulating blood cells. Most mediate their responses via G proteins, although the P2X receptors are a subfamily of ion-gated ion channels (Burnstock, 2000). P2Y receptors have been found to activate MAP kinase (Neary, 2000). Modulation of Vascular Responses by Endothelium-Derived Factors Furchgott and colleagues demonstrated that an intact endothelium was necessary to achieve vascular relaxation in response to ACh (see Furchgott, 1984, 1999). This inner layer of the blood vessel now is known to modulate autonomic and hormonal effects on the contractility of blood vessels. In response to a variety of vasoactive agents and even physical stimuli, the endothelial cells release a short-lived vasodilator called endothelium-derived relaxing factor (EDRF), now known to be nitric oxide. Less commonly an endothelium-derived hyperpolarizing factor (EDHF) and endothelium-derived contracting factor (EDCF) of as yet undefined compositions are released (Vanhoutte, 1996). EDCF formation is dependent on cyclooxygenase activity. Products of inflammation and platelet aggregation such as serotonin, histamine, bradykinin, purines, and thrombin exert all or part of their action by stimulating the release of nitric oxide. Endothelial cell-dependent mechanisms of relaxation are important in a variety of vascular beds, including the coronary circulation (Hobbs et al., 1999). Activation of specific G proteinlinked receptors on endothelial cells promotes release of nitric oxide. Nitric oxide diffuses readily to the underlying smooth muscle and induces relaxation of vascular smooth muscle by activating guanylyl cyclase, which increases cyclic GMP concentrations. Nitrovasodilating drugs used to lower blood pressure or to treat ischemic heart disease probably act through conversion to or release of nitric oxide (see Chapter 32: Drugs Used for the Treatment of Myocardial Ischemia). Nitric oxide also has been shown to be released from certain nerves (nitrergic) innervating blood vessels. It has a negative inotropic action on the heart. Recently, it has become clear that alterations in the release or
action of nitric oxide may have importance for a number of major clinical
situations such as atherosclerosis (Hobbs et al., 1999; Ignarro, 1999).
Furthermore, there is evidence suggesting that the hypotension of endotoxemia
or that induced by cytokines is mediated at least in part by induction of
enhanced release of nitric oxide; consequently, increased release of nitric
oxide may have pathological significance in septic shock. Nitric oxide is
synthesized from L-arginine and
molecular oxygen by nitric oxide synthase (NOS). There are three known
forms of this enzyme (Moncada et al., 1997). One form is constitutive,
residing in the endothelial cell and releasing nitric oxide over short
periods in response to receptor-mediated increases in cellular Ca2+
(eNOS) (Michel and Feron, 1997). A second form is responsible for the Ca2+-dependent
release from neurons (nNOS). The third form of NOS is induced after activation
of cells by cytokines and bacterial endotoxins and, once expressed,
synthesizes nitric oxide for long periods of time (iNOS). This Ca2+-independent,
high-output form is responsible for the above-mentioned toxic manifestations
of nitric oxide. Glucocorticoids inhibit the expression of inducible, but not
constitutive, forms of nitric oxide synthase in vascular endothelial cells.
However, other endothelium-derived factors also may be involved in
vasodilation and hyperpolarization of the smooth muscle cell. There has been
considerable interest in the possibility that NOS inhibitors might have
therapeutic benefit, for example, in septic shock and neurodegenerative
diseases ( Full contractile responses of cerebral arteries also require an intact endothelium. A family of peptides, termed endothelins, are stored in vascular endothelial cells. Their release onto smooth muscle promotes contraction by stimulation of endothelin receptors. Endothelins contribute to the maintenance of vascular homostasis by acting via multiple endothelin receptors (Sokolovsky, 1995) to reverse the response to nitric oxide (Rubanyi and Polokoff, 1994). |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 6716
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved
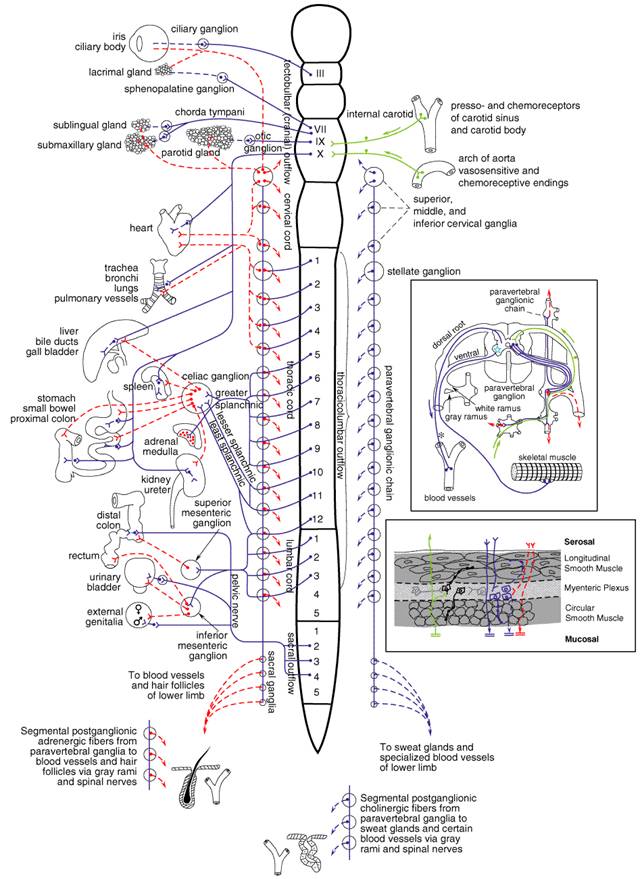

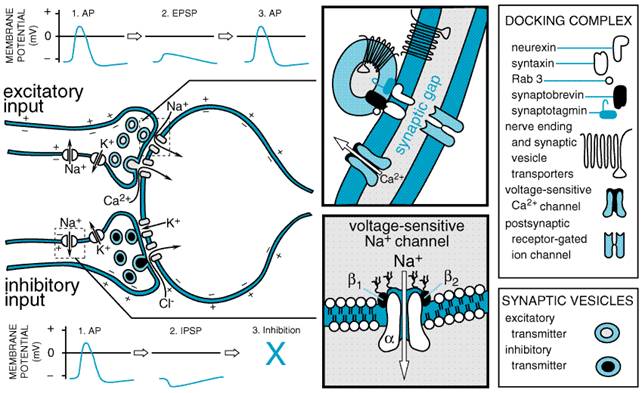
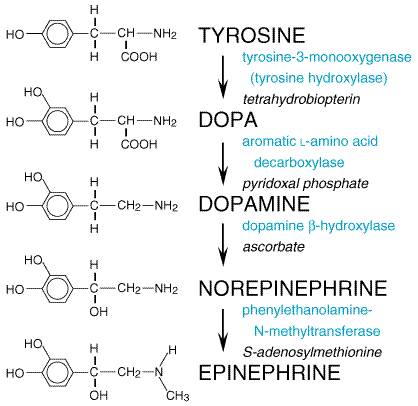 ydroxylation of
tyrosine generally is regarded as the rate-limiting step in the biosynthesis
of catecholamines (Zigmond et al., 1989), and tyrosine hydroxylase is
activated following stimulation of adrenergic nerves or the adrenal medulla.
The enzyme is a substrate for cyclic AMPdependent and Ca2+-calmodulin-sensitive
protein kinase and protein kinase C; kinase-catalyzed phosphorylation may be
associated with increased hydroxylase activity (Zigmond et al., 1989; Daubner
et al., 1992). This is an important acute mechanism for increasing
catecholamine synthesis in response to increased nerve stimulation. In
addition, there is a delayed increase in tyrosine hydroxylase gene expression
after nerve stimulation. There is evidence suggesting that this increased
expression can occur at multiple levels of regulation, including
transcription, RNA processing, regulation of RNA stability, translation, and
enzyme stability (Kumer and Vrana, 1996). These mechanisms serve to maintain
the content of catecholamines in response to increased release of these
transmitters. In addition, tyrosine hydroxylase is subject to feedback
inhibition by catechol compounds, an allosteric modulation of enzyme activity.
Patients with mutations in the tyrosine hydroxylase gene have been described
(Wevers et al., 1999).
ydroxylation of
tyrosine generally is regarded as the rate-limiting step in the biosynthesis
of catecholamines (Zigmond et al., 1989), and tyrosine hydroxylase is
activated following stimulation of adrenergic nerves or the adrenal medulla.
The enzyme is a substrate for cyclic AMPdependent and Ca2+-calmodulin-sensitive
protein kinase and protein kinase C; kinase-catalyzed phosphorylation may be
associated with increased hydroxylase activity (Zigmond et al., 1989; Daubner
et al., 1992). This is an important acute mechanism for increasing
catecholamine synthesis in response to increased nerve stimulation. In
addition, there is a delayed increase in tyrosine hydroxylase gene expression
after nerve stimulation. There is evidence suggesting that this increased
expression can occur at multiple levels of regulation, including
transcription, RNA processing, regulation of RNA stability, translation, and
enzyme stability (Kumer and Vrana, 1996). These mechanisms serve to maintain
the content of catecholamines in response to increased release of these
transmitters. In addition, tyrosine hydroxylase is subject to feedback
inhibition by catechol compounds, an allosteric modulation of enzyme activity.
Patients with mutations in the tyrosine hydroxylase gene have been described
(Wevers et al., 1999).