| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Vasopressin and Other Agents Affecting the Renal Conservation of Water
Overview
|
Precise regulation of body fluid osmolality is essential. It is controlled by a finely tuned, intricate homeostatic mechanism that operates by adjusting both the rate of water intake and the rate of solute-free water excretion by the kidneysi.e., water balance. Abnormalities in this homeostatic system can be caused by genetic diseases, acquired diseases, or drugs and may result in serious and potentially life-threatening deviations in plasma osmolality. The goals of this chapter are to describe the physiological mechanisms that regulate plasma osmolality, to discuss the diseases that perturb those mechanisms, and to examine pharmacological approaches for treating disorders of water balance. Argininevasopressin (the antidiuretic hormone in human beings) is the main hormone involved in regulation of body fluid osmolality. Many diseases of water homeostasis and many pharmacological strategies for correcting such disorders pertain to vasopressin. Accordingly, vasopressin is the major focus of this chapter and is discussed with regard to: (1) chemistry (including the chemistry of vasopressin agonists and antagonists); (2) physiology (including anatomical considerations; the synthesis, transport, and storage of vasopressin and the regulation of vasopressin secretion); (3) basic pharmacology (including vasopressin receptors and their signal transduction pathways, renal actions of vasopressin, pharmacological modification of the antidiuretic response to vasopressin, and nonrenal actions of vasopressin); (4) diseases affecting the vasopressin system (diabetes insipidus, syndrome of inappropriate secretion of antidiuretic hormone, and other water-retaining states); and (5) clinical pharmacology of vasopressin peptides (therapeutic uses, pharmacokinetics, toxicities, adverse effects, contraindications, and drug interactions). A small number of other drugs can be used to treat abnormalities of water balance; a discussion of these agents is integrated into the section on diseases affecting the vasopressin system. |
Introduction to Vasopressin
|
Immunoreactive vasopressin has been observed in neurons from organisms belonging to the first animal phylum with a nervous system (e.g., Hydra attenuata), and vasopressin-like peptides have been isolated and characterized from both mammalian and nonmammalian vertebrates as well as from invertebrates (Table 301). Genes encoding vasopressin-like peptides probably evolved more than 700 million years ago. With the emergence of life on land, vasopressin became the mediator of a remarkable regulatory system for the conservation of water. The hormone is released by the posterior pituitary whenever water deprivation causes an increased plasma osmolality or whenever the cardiovascular system is challenged by hypovolemia and/or hypotension. In amphibians, the target organs for vasopressin are skin and the urinary bladder, whereas in other vertebrates, including human beings, the site of action is primarily the renal collecting duct. In each of these target tissues, vasopressin acts by increasing the permeability of the cell membrane to water, thus permitting water to move passively down an osmotic gradient across skin, bladder, or collecting duct into the extracellular compartment. In view of the long evolutionary history of vasopressin, it is not surprising that vasopressin acts at sites in the nephron other than the collecting duct and on tissues other than the kidney. Vasopressin is a potent vasopressor, and its name was originally chosen in recognition of its vasoconstrictor action. Vasopressin is a neurotransmitter; among its actions in the central nervous system (CNS) are apparent roles in the secretion of adrenocorticotropic hormone (ACTH) and in the regulation of the cardiovascular system, temperature, and other visceral functions. Vasopressin also promotes the release of coagulation factors by the vascular endothelium and increases platelet aggregability; therefore, it may play a role in hemostasis. |
Chemistry of Vasopressin Receptor Agonists and Antagonists
|
Chemistry of Vasopressin Receptor Agonists du Vigneaud and coworkers (1954) determined the structures of vasopressin and oxytocin and accomplished the complete synthesis of each. A number of vasopressin-like peptides occur naturally (Table 301). All are nonapeptides; contain cysteine residues in positions 1 and 6; have an intramolecular disulfide bridge between the two cysteine residues (essential for agonist activity); have additional conserved amino acids in positions 5, 7, and 9 (asparagine, proline, and glycine, respectively); contain a basic amino acid in position 8; and are amidated on the carboxyl terminus. In all mammals except swine, the neurohypophyseal peptide is 8-arginine vasopressin, and the terms vasopressin, arginine vasopressin (AVP), and antidiuretic hormone (ADH) are used interchangeably. The chemical structure of oxytocin is closely related to that of vasopressin, i.e., oxytocin is [Ile3, Leu8]AVP. Oxytocin binds to specific oxytocin receptors on myoepithelial cells in the mammary gland and on smooth muscle cells in the uterus, causing milk ejection and uterine contraction, respectively. Inasmuch as vasopressin and oxytocin are structurally similar, it is not surprising that vasopressin and oxytocin agonists and antagonists can bind to each other's receptors. Therefore, most of the available peptide vasopressin agonists and antagonists have some affinity for oxytocin receptors; at high doses, they may block or mimic the effects of oxytocin (Manning and Sawyer, 1989). With the advent of solid-phase peptide synthesis, many vasopressin analogs were synthesized with the goal of increasing duration of action and selectivity for vasopressin receptor subtypes (V1versus V2 vasopressin receptors, which mediate pressor responses and antidiuretic responses, respectively). In 1967, Zaoral and coworkers announced the synthesis of desmopressin: 1-deamino-8-D-arginine vasopressin (DDAVP) (Table 301). Deamination at position 1 increases duration of action and increases antidiuretic activity without increasing vasopressor activity. Substitution of D-arginine for L-arginine greatly reduces vasopressor activity without reducing antidiuretic activity. Thus, the antidiuretic-to-vasopressor ratio for desmopressin is approximately 3000-fold greater than that for vasopressin, and desmopressin now is the preferred drug for the treatment of central diabetes insipidus (Robinson, 1976). Substitution of valine for glutamine in position 4 further increases the antidiuretic selectivity, and the antidiuretic-to-vasopressor ratio for deamino[Val4, D-Arg8] AVP (Table 301) is approximately 11,000-fold greater than that for vasopressin. Increasing V1 selectivity has proved more difficult than increasing V2 selectivity (Thibonnier, 1990). However, a limited number of agonists have been developed with modest selectivity for V1 receptors (seeTable 301). Vasopressin receptors in the adenohypophysis that mediate vasopressin-induced ACTH release are neither classical V1 nor V2 receptors. Since the vasopressin receptors in the adenohypophysis appear to share a common signal transduction mechanism with classical V1 receptors, and since many vasopressin analogs with vasoconstrictor activity release ACTH, V1 receptors have been subclassified into V1a (vascular/hepatic) and V1b (pituitary) receptors (Jard et al., 1986). Vasopressin agonists selective for both V1a (Thibonnier, 1990) and V1b receptors (Schwartz et al., 1991) have been described (seeTable 301). Chemistry of Vasopressin Receptor Antagonists The impetus for the development of specific vasopressin receptor antagonists is the belief that such drugs may be useful in a number of clinical settings. Selective V1a antagonists may be beneficial when total peripheral resistance is increased (e.g., congestive heart failure and hypertension); selective V2 antagonists could be useful whenever reabsorption of solute-free water is excessive (e.g., the syndrome of inappropriate secretion of antidiuretic hormone and hyponatremia associated with a reduced effective blood volume). Combined V1a/V2 receptor antagonists might be beneficial in diseases associated with a combination of increased peripheral resistance and dilutional hyponatremia (e.g., congestive heart failure). Shortly after the synthesis of vasopressin, du Vigneaud and coworkers
began designing antagonists of vasopressin's pharmacological effects. Since
that time, numerous vasopressin receptor antagonists have been synthesized (Manning
et al., 1993; Lszlet al., 1991). Highly selective V1
and V2 peptide antagonists that are structural analogs of
vasopressin have been synthesized (seeTable 302 for examples),
including both cyclic and linear peptides. [1-( |
Anatomy
The antidiuretic mechanism in mammals involves two anatomical components: a CNS component for the synthesis, transport, storage, and release of vasopressin, and a renal collecting duct system composed of epithelial cells that respond to vasopressin by increasing their permeability to water. The relevant anatomy of the renal collecting duct system is described in Chapter 29: Diuretics. The CNS component of the antidiuretic mechanism is called the hypothalamiconeurohypophyseal system and consists of neurosecretory neurons with perikarya located predominantly in two specific hypothalamic nuclei, the supraoptic nucleus (SON) and the paraventricular nucleus (PVN). The long axons of neurons in the SON and PVN traverse the supraopticohypophyseal tract to terminate in the median eminence and pars nervosa of the posterior pituitary.
Synthesis
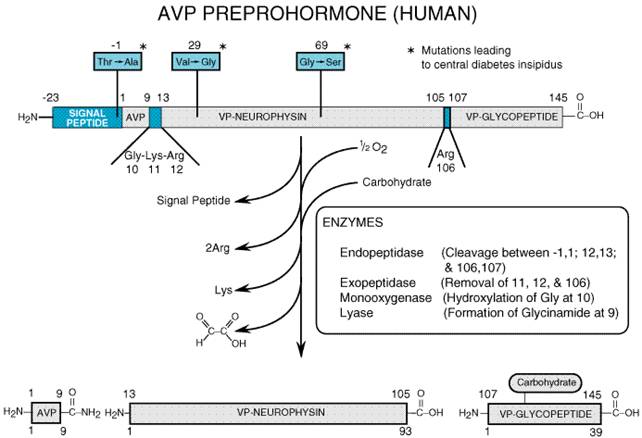
Vasopressin and oxytocin are synthesized in the perikarya of magnocellular neurons in the SON and PVN; the two hormones are synthesized predominantly in separate neurons. Vasopressin synthesis appears to be regulated solely at the transcriptional level (Robinson and Fitzsimmons, 1993), and the molecular mechanism of vasopressin synthesis has been elucidated in considerable detail (Archer, 1993). In human beings, a 168-amino-acid preprohormone (Figure 301) is synthesized, and a signal peptide (residues -23 to -1) assures incorporation of the nascent polypeptide into ribosomes. During synthesis, the signal peptide is removed to form the vasopressin prohormone, and vesicle-mediated translocations maneuver the prohormone through the rough endoplasmic reticulum and cis-, medial-, and trans-Golgi compartments, so that the prohormone emerges incorporated into large (0.1- to 0.3-micron) membrane-enclosed granules. The prohormone consists of three domains: vasopressin (residues 1 to 9), vasopressin (VP)neurophysin (residues 13 to 105), and VPglycopeptide (residues 107 to 145). The vasopressin domain is linked to the VPneurophysin domain through a glycinelysinearginine processing signal, and the VPneurophysin domain is linked to the VPglycopeptide domain by an arginine-processing signal. In the secretory granules, an endopeptidase, exopeptidase, monooxygenase, and lyase act sequentially on the prohormone to produce vasopressin, VPneurophysin (sometimes referred to as neurophysin II or MSELneurophysin), and VPglycopeptide (sometimes called copeptin). The synthesis and transport of vasopressin are dependent on the conformation of the preprohormone. In particular, VPneurophysin binds vasopressin and is critical to the correct processing, transport, and storage of vasopressin (Breslow, 1993). Genetic mutations in either the signal peptide or VPneurophysin give rise to central diabetes insipidus (Raymond, 1994).
|
Figure 301. Processing of the 168Amino Acid Human 8-Arginine Vasopressin (AVP) Preprohormone to AVP, Vasopressin (VP)Neurophysin, and VPGlycopeptide. |
Transport and Storage
The process of axonal transport of vasopressin-containing granules is rapid, and newly synthesized neurohypophyseal hormones arrive at the posterior lobe within 30 minutes of a stimulus. The axons involved in transport of granules have two destinations, carrying vasopressin not only to classical storage sites in the neurohypophysis but also to the external zone of the median eminence, where vasopressin enters the adenohypophyseal portal circulation and plays a role as a corticotropin-releasing factor.
Maximal release of vasopressin occurs when impulse frequency is approximately 12 spikes per second for 20 seconds. Higher frequencies or longer periods of stimulation lead to diminished hormone release (fatigue). Appropriately, vasopressin-releasing cells demonstrate an atypical pattern of spike activity characterized by rapid phasic bursts (5 to 12 spikes per second for 15 to 60 seconds) separated by quiescent periods (15 to 60 seconds in duration). This pattern is orchestrated by activation and inactivation of ion channels in the magnocellular neurons and provides for optimal release of vasopressin (Leng et al., 1992).
Vasopressin Synthesis Outside of the CNS
Vasopressin also is synthesized by the heart (Hupf et al., 1999) and adrenal gland (Guillon et al., 1998). In the heart, elevated wall stress increases vasopressin synthesis several-fold. Cardiac synthesis of vasopressin is predominantly vascular and perivascular and may contribute to impaired ventricular relaxation and coronary vasoconstriction. Vasopressin synthesis in the adrenal medulla stimulates catecholamine secretion from chromaffin cells and may promote adrenal cortical growth and stimulate aldosterone synthesis.
Regulation of Vasopressin Secretion
An increase in plasma osmolality is the principal physiological stimulus for vasopressin secretion. Severe hypovolemia/hypotension also is a powerful stimulus for vasopressin release. In addition, pain, nausea, and hypoxia can stimulate vasopressin secretion, and several endogenous hormones and pharmacological agents can modify vasopressin release.
Hyperosmolality
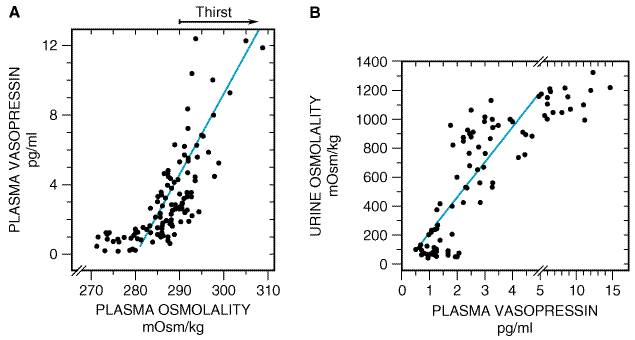
The relationship between plasma osmolality and plasma vasopressin concentration is shown in Figure 302A, and the relationship between plasma vasopressin levels and urine osmolality is illustrated in Figure 302B. The osmolality threshold for secretion is approximately 280 mOsm/kg. Below the threshold, vasopressin is barely detectable in plasma, and above the threshold, vasopressin levels are a steep and linear function of plasma osmolality. In fact, a 2% elevation in plasma osmolality causes a two- to threefold increase in plasma vasopressin levels. Therefore, a small increase in plasma osmolality leads to enhanced vasopressin secretion, which, in turn, causes increased solute-free water reabsorption (as evidenced by the increased urine osmolality). Increases in plasma osmolality (due to insensible water losses) above 290 mOsm/kg lead to an intense desire for water (thirst). Thus, the vasopressin system affords the organism longer thirst-free periods and, in the event that water is unavailable, allows the organism to survive longer periods of water deprivation. It is important to point out, however, that above a plasma osmolality of approximately 290 mOsm/kg, plasma levels of vasopressin exceed 5 pM. Since urinary concentration is maximal (about 1200 mOsm/kg) when vasopressin levels exceed 5 pM, further defense against hypertonicity is entirely dependent on water intake rather than on decreases in water loss.
|
Figure 302. A. The relationship between plasma osmolality and plasma vasopressin levels. Plasma osmolality associated with thirst is indicated by arrow. B. The relationship between plasma vasopressin levels and urine osmolality. (From Robertson et al., 1977, and Kovacs and Robertson, 1992, with permission.) |
Several CNS structures are involved in osmotic stimulation of vasopressin release; these structures are collectively referred to as the osmoreceptive complex. Although magnocellular neurons in the SON and PVN are osmosensitive, afferent inputs from other components of the osmoreceptive complex are required for a normal vasopressin response. The SON and PVN receive projections from the subfornical organ (SFO) and the organum vasculosum of the lamina terminalis (OVLT) either directly or indirectly [via the median preoptic nucleus (MnPO)]. Subgroups of neurons in the SFO, OVLT, and MnPO are either osmoreceptors or osmoresponders (i.e., are stimulated by osmoreceptive neurons located at other sites). Thus, a web of interconnecting neurons contributes to osmotically induced vasopressin secretion.
Aquaporin 4, a water-selective channel, is associated with CNS structures involved in osmoregulation and may confer osmosensitivity. In the CNS, aquaporin 4 resides on glial and ependymal cells rather than neurons, suggesting that osmotic status may be communicated to neuronal cell by a glialneuron interaction (Wells, 1998).
Hypovolemia and Hypotension
Vasopressin secretion also is regulated hemodynamically by changes in effective blood volume and/or arterial blood pressure (Robertson, 1992). Reductions in effective blood volume and/or arterial blood pressure, regardless of the cause (e.g., hemorrhage, sodium depletion, diuretics, heart failure, hepatic cirrhosis with ascites, adrenal insufficiency, hypotensive drugs), may be associated with high circulating concentrations of vasopressin. However, unlike osmoregulation, hemodynamic regulation of vasopressin secretion is exponential, i.e., small decreases (5% to 10%) in blood volume and/or pressure have little effect on vasopressin secretion, whereas larger decreases (20% to 30%) can increase vasopressin levels to 20 to 30 times normal levels (exceeding the concentration of vasopressin required to induce maximal antidiuresis). Vasopressin is one of the most potent vasoconstrictors known, and the vasopressin response to hypovolemia or hypotension serves as a mechanism to stave off cardiovascular collapse during periods of severe blood loss and/or hypotension. Importantly, hemodynamic regulation of vasopressin secretion does not disrupt osmotic regulation; rather, hypovolemia/hypotension alters the setpoint and slope of the plasma osmolalityplasma vasopressin relationship (Figure 303).
|
Figure 303. Interactions between Osmolality and Hypovolemia/Hypotension. Numbers in circles refer to percentage increase (+) or decrease () in blood volume or arterial blood pressure. N indicates normal blood volume/blood pressure. (From Robertson, 1992, with permission.) |
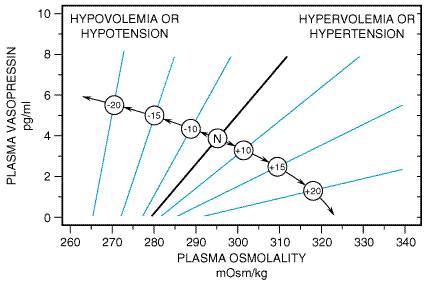
The neuronal pathways that mediate hemodynamic regulation of vasopressin release are completely different from those involved in osmoregulation. Baroreceptors in the left atrium, left ventricle, and pulmonary veins sense blood volume (filling pressures), and baroreceptors in the carotid sinus and aorta monitor arterial blood pressure. Nerve impulses reach brainstem nuclei predominantly through the vagus and glossopharyngeal nerves; these signals are relayed to the nucleus of the solitary tract, then to the A1-noradrenergic cell group in the caudal ventrolateral medulla, and finally to the SON and PVN (Cunningham and Sawchenko, 1991).
Hormones and Neurotransmitters
There is a large, sometimes contradictory, body of literature on modulation of vasopressin secretion by hormones and neurotransmitters (Renaud and Bourque, 1991). Vasopressin-synthesizing magnocellular neurons have a large array of receptors on both perikarya and nerve terminals; therefore, vasopressin release can be accentuated or attenuated by chemical agents acting at both ends of the magnocellular neuron. Also, hormones and neurotransmitters can modulate vasopressin secretion by stimulating or inhibiting neurons in nuclei that project, either directly or indirectly, to the SON and PVN. Because of these complexities, the results of any given investigation may depend critically on the route of administration of the agent and on the experimental paradigm. In many cases, the precise mechanism by which a given agent modulates vasopressin secretion is either unknown or controversial, and the physiological relevance of modulation of vasopressin secretion by most hormones and neurotransmitters is unclear.
Nonetheless, several agents are known to stimulate vasopressin
secretion, including acetylcholine (via nicotinic receptors), histamine
(via H1 receptors), dopamine (via both D1
and D2 receptors), glutamine, aspartate, cholecystokinin,
neuropeptide Y, substance P, vasoactive intestinal polypeptide, prostaglandins,
and angiotensin II. Inhibitors of vasopressin secretion include atrial
natriuretic peptide, gamma-aminobutyric acid, and opioids (particularly
dynorphin via![]() receptors). Of all the
aforementioned hormones/neurotransmitters, angiotensin II has received the most
attention (Phillips, 1987). Angiotensin II, when applied directly to
magnocellular neurons in the SON and PVN, increases neuronal excitability; when
applied to the MnPO, angiotensin II indirectly stimulates magnocellular neurons
in the SON and PVN. In addition, angiotensin II stimulates
angiotensin-sensitive neurons in the OVLT and SFO (circumventricular nuclei
lacking a bloodbrain barrier) that project to the SON/PVN. Thus, both
angiotensin II synthesized in the brain and that formed in the circulation may
stimulate vasopressin release. Inhibition of the conversion of angiotensin II
to angiotensin III blocks angiotensin IIinduced vasopressin release,
suggesting that angiotensin III is the main effector peptide of the brain
reninangiotensin system controlling vasopressin release (Zini et al.,
1996).
receptors). Of all the
aforementioned hormones/neurotransmitters, angiotensin II has received the most
attention (Phillips, 1987). Angiotensin II, when applied directly to
magnocellular neurons in the SON and PVN, increases neuronal excitability; when
applied to the MnPO, angiotensin II indirectly stimulates magnocellular neurons
in the SON and PVN. In addition, angiotensin II stimulates
angiotensin-sensitive neurons in the OVLT and SFO (circumventricular nuclei
lacking a bloodbrain barrier) that project to the SON/PVN. Thus, both
angiotensin II synthesized in the brain and that formed in the circulation may
stimulate vasopressin release. Inhibition of the conversion of angiotensin II
to angiotensin III blocks angiotensin IIinduced vasopressin release,
suggesting that angiotensin III is the main effector peptide of the brain
reninangiotensin system controlling vasopressin release (Zini et al.,
1996).
Pharmacological Agents
A number of drugs alter the osmolality of urine. It has been hypothesized that the action of many agents involves stimulation or inhibition of the secretion of vasopressin (Robertson, 1992). In some cases, the mechanism by which a drug alters vasopressin secretion involves direct effects on one or more CNS structures involved in the regulation of vasopressin secretion. In other cases, vasopressin secretion is indirectly altered by the effects of a drug on blood volume, arterial blood pressure, pain, or nausea. In most cases, the mechanism is not known. Stimulators of vasopressin secretion include vincristine, cyclophosphamide, tricyclic antidepressants, nicotine, epinephrine, and high doses of morphine. Lithium, which inhibits the renal effects of vasopressin, also enhances vasopressin secretion. Inhibitors of vasopressin secretion include ethanol, phenytoin, low doses of morphine, glucocorticoids, fluphenazine, haloperidol, promethazine, oxilorphan, and butorphanol. Carbamazepine has a renal action to produce antidiuresis in patients with central diabetes insipidus but actually inhibits vasopressin secretion via a central action.
Basic Pharmacology of Vasopressin
|
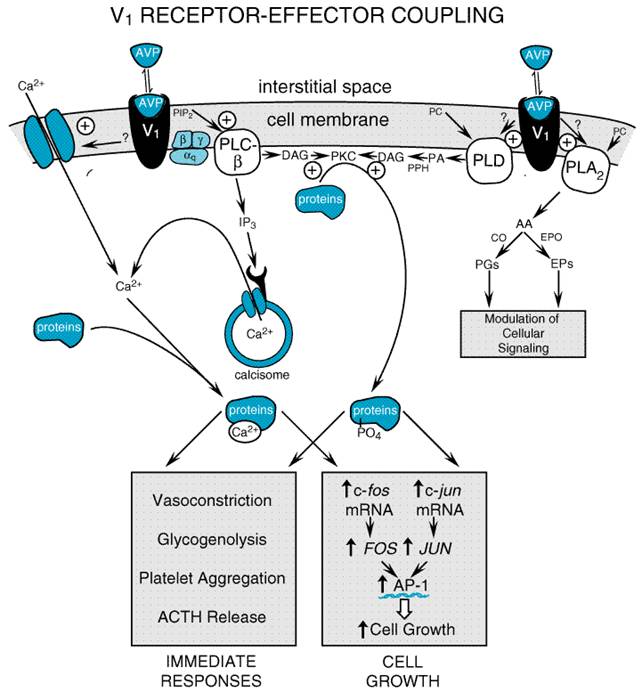
Vasopressin Receptors The cellular effects of vasopressin are mediated by interactions of the hormone with the two principal types of receptors, V1 and V2. V1 receptors have been subclassified further as V1a and V1b. The V1a receptor is the most widespread subtype of vasopressin receptor; it is found in vascular smooth muscle, the adrenal gland, myometrium, the bladder, adipocytes, hepatocytes, platelets, renal medullary interstitial cells, vasa recta in the renal microcirculation, epithelial cells in the renal cortical collecting duct, spleen, testis, and in many CNS structures. The adenohypophysis and the adrenal medulla are known to contain V1b receptors, whereas V2 receptors are located predominantly in principal cells of the renal collecting duct system. Although originally defined by pharmacological criteria, V1a (Morel et al., 1992), V1b (Sugimoto et al., 1994), and V2 receptors (Birnbaumer et al., 1992; Lolait et al., 1992) have been cloned, and vasopressin receptors now are defined by their primary amino acid sequences. The cloned vasopressin receptors are typical G proteincoupled receptors containing seven transmembrane-spanning domains. Manning and coworkers (1999) have synthesized novel, hypotensive vasopressin peptide agonists that do not interact with V1a, V1b, or V2 receptors and may stimulate a putative vasopressin vasodilatory receptor. V1 ReceptorEffector Coupling Considerable progress has been made in defining the mechanisms by
which vasopressin receptors are coupled to biological responses (Thibonnier et
al., 1993; Holtzman and Ausiello, 1994). Figure 304 summarizes the
current model of V1 receptoreffector coupling. When vasopressin
binds to V1 receptors, a G proteinmediated activation of several
membrane-bound phospholipases ensues. Activation of phospholipase C-
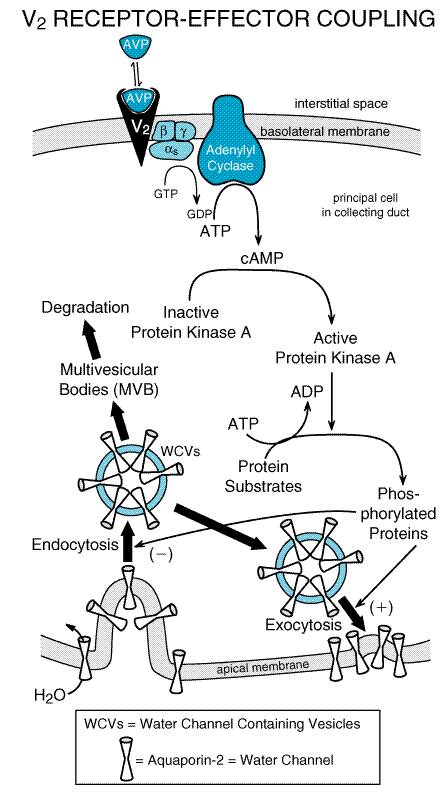
V2 ReceptorEffector Coupling Principal cells in the renal collecting duct have V2 receptors on their basolateral membranes that are coupled to adenylyl cyclase via the timulatory G protein Gs (seeFigure 305). Consequently, when vasopressin binds to V2 receptors, adenylyl cyclase is stimulated, and intracellular levels of cyclic AMP are increased. Activation of cyclic AMPdependent protein kinase (protein kinase A) mediates the hydroosmotic effects of vasopressin (Snyder et al., 1992) via protein phosphorylation. In this regard, protein kinase Amediated protein phosphorylation triggers an increased rate of exocytosis of water channelcontaining vesicles (WCVs) into the apical membrane and a decreased rate of endocytosis of WCVs from the apical membrane. The distribution of WCVs between the cytosolic compartment and the apical membrane compartment is thus shifted in favor of the apical membrane compartment (Knepper and Nielsen, 1993; Nielsen et al., 1999). Because WCVs contain preformed, functional water channels (aquaporin 2) their increased rate of insertion into and decreased rate of removal from the apical membrane greatly increases the water permeability of the apical membrane.
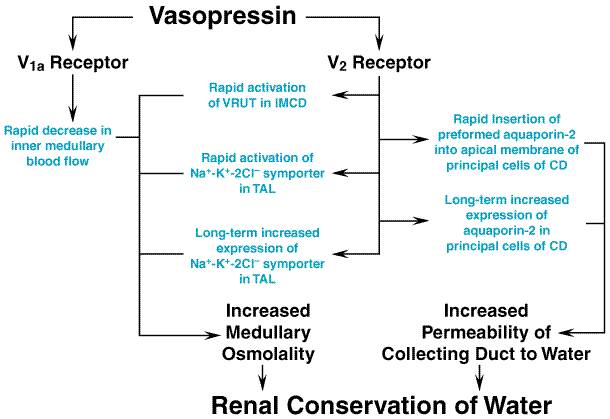
Aquaporins are a family of water channel proteins that allow water molecules to cross biological membranes (Nielsen et al., 1999; Marples et al., 1999). Aquaporins have six membrane domains connected by five loops (A to E). Loops B and E dip into the cell membrane, and the asparagine-proline-alanine sequences in each B and E loop interact to form a water pore. Aquaporins generally form tetrameric complexes in cell membranes. Of the nine cloned mammalian aquaporins, six are found in the kidney. Aquaporin 1 is present in the apical and basolateral membrane of the proximal tubule and in the thin descending limb. Aquaporin 2 resides in the apical membrane and WCVs of the collecting duct principal cells, whereas aquaporins 3 and 4 are present in the basolateral membrane of principal cells. Aquaporin 7 is in the apical brush border of the straight proximal tubule. Although aquaporin 6 is found in the kidney, its distribution is unknown. Aquaporin 2, the water channel in WCVs, is phosphorylated on serine 256 by protein kinase A, and this is the first step leading to vasopressin-induced insertion of WCVs into apical membranes (Nishimoto et al., 1999). In addition to increasing the insertion of aquaporin 2 into apical membranes in collecting duct principal cells, vasopressin also increases the expression of aquaporin 2 mRNA and protein (Marples et al., 1999). This effect is mediated by protein kinase A phosphorylation of cyclic AMPresponse element binding protein (CREB). Phosphorylated CREB is a transcription factor that binds to the cyclic AMPresponse element (CRE) in the 5'-untranslated region of the gene encoding aquaporin 2 and increases its transcription. Thus, chronic dehydration leads to long-term upregulation of aquaporin 2 and water transport in the collecting duct. For maximum concentration of urine, large amounts of urea must be deposited in the interstitium of the inner medullary collecting duct. It is not surprising, therefore, that V2 receptor activation also increases urea permeability by 400% in the terminal portions of the inner medullary collecting duct. The mechanism by which V2 receptors increase urea permeability involves activation of a vasopressin-regulated urea transporter (termed VRUT, UT1, or UT-A1), most likely by protein kinase Ainduced phosphorylation (Star et al., 1988; Sands, 1999). The kinetics of vasopressin-induced water and urea permeability are different (Nielsen and Knepper, 1993), and vasopressin-induced regulation of VRUT does not entail vesicular trafficking to the plasma membrane (Inoue et al., 1999). In addition to increasing the permeability of the collecting duct to water and the permeability of the inner medullary collecting duct to urea, V2 receptor activation also increases both short- and long-term NaCl transport in the thick ascending limb. Increased transport in the thick ascending limb augments the countercurrent multiplication system and thereby increases the osmolality of the medullary interstitium and the reabsorption of water (Knepper et al., 1999). This effect, which is most likely mediated by cyclic AMP and protein kinase Ainduced phosphorylation, involves the immediate stimulation of Na+K+2Cl symporter expression (Kim et al., 1999). The multiple mechanisms by which vasopressin increases water reabsorption are summarized in Figure 306.
Renal Actions of Vasopressin There are several sites of vasopressin action in the kidney involving both V1 and V2 receptors. V1 receptors mediate contraction of mesangial cells in the glomerulus and contraction of vascular smooth muscle cells in the vasa recta and efferent arteriole (Edwards et al., 1989). Indeed, V1 receptormediated reduction in inner medullary blood flow contributes to the maximum concentrating capacity of the kidney (Franchini and Cowley, 1996) (seeFigure 306). V1 receptors also stimulate prostaglandin synthesis by medullary interstitial cells. Since prostaglandin E2 inhibits adenylyl cyclase in the collecting duct, stimulation of prostaglandin synthesis by V1 receptors may function to restrain V2 receptormediated antidiuresis (Sonnenberg and Smith, 1988). V1 receptors on principal cells in the cortical collecting duct (Burnatowska-Hledin and Spielman, 1989) may directly inhibit V2 receptormediated water flux via activation of protein kinase C (Schlondorff and Levine, 1985). Without question, V2 receptors mediate the most prominent response to vasopressin, i.e., increased water permeability of the collecting duct. Indeed, vasopressin can increase water permeability in the collecting duct at concentrations as low as 50 fM. Thus, V2 receptor-mediated effects of vasopressin occur at concentrations far lower than are required to engage the V1 receptor-mediated actions; however, this differential sensitivity may not be due to differences in receptor affinities, since cloned rat V1a and V2 receptors have similar affinities for vasopressin (Kd= 0.7 and 0.4 nM, respectively). The differential sensitivity may instead be due to differential amplification of signal transduction pathways evoked by these receptors. The collecting duct system is critical for the conservation of water. By the time tubular fluid arrives at the cortical collecting duct, it has been rendered hypotonic by the upstream diluting segments of the nephron that reabsorb NaCl without reabsorbing water. In the well-hydrated subject, plasma osmolality is in the normal range, concentrations of vasopressin are low, the entire collecting duct is relatively impermeable to water, and the urine is dilute. Under conditions of dehydration, plasma osmolality is increased, concentrations of vasopressin are elevated, and the collecting duct becomes permeable to water. The osmotic gradient between the dilute tubular urine and the hypertonic renal interstitial fluid (which becomes progressively more hypertonic in deeper regions of the renal medulla) provides for the osmotic flux of water out of the collecting duct. The final osmolality of urine may be as high as 1200 mOsm/kg in human beings, and a significant saving of solute-free water is thus possible. Other renal actions mediated by V2 receptors include increased urea transport in the inner medullary collecting duct and increased NaCl transport in the thick ascending limb; both effects contribute to the urine-concentrating ability of the kidney. V2 receptors also increase Na+ transport in the cortical collecting duct (Schafer and Troutman, 1990). Pharmacological Modification of the Antidiuretic Response to Vasopressin Nonsteroidal antiinflammatory drugs (NSAIDs) (seeChapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout), particularly indomethacin, enhance the antidiuretic response to vasopressin. Since prostaglandins attenuate antidiuretic responses to vasopressin and NSAIDs inhibit prostaglandin synthesis, reduced prostaglandin production probably accounts for the potentiation of vasopressin's antidiuretic response. Other drugs that enhance the antidiuretic effects of vasopressin include carbamazepine and chlorpropamide; however, the mechanisms by which these agents potentiate the antidiuretic response to vasopressin are not known. In rare instances, chlorpropamide can induce water intoxication. A number of drugs inhibit the antidiuretic actions of vasopressin. Lithium is of particular importance because of its wide use in the treatment of manic-depressive disorders. Lithium-induced polyuria is usually, but not always, reversible (Ramsey and Cox, 1982). Acutely, lithium appears to reduce V2 receptormediated stimulation of adenylyl cyclase. The mechanism of this effect may involve attenuation of Gs-mediated activation of adenylyl cyclase (Cogan and Abramow, 1986; Goldberg et al., 1988) and/or enhancement of Gi-mediated inhibition of adenylyl cyclase (Yamaki et al., 1991). Also, lithium increases plasma levels of parathyroid hormone, and parathyroid hormone is a partial antagonist to vasopressin (Carney et al., 1996). In most patients, the antibiotic demeclocycline attenuates the antidiuretic effects of vasopressin, and this action of demeclocycline is probably due to decreased accumulation and action of cyclic AMP (Singer and Rotenberg, 1973). Nonrenal Actions of Vasopressin Vasopressin and related peptides are ancient hormones in evolutionary terms, and they are found in species that do not concentrate urine. It is thus not surprising that vasopressin has nonrenal actions in mammals. Cardiovascular System The cardiovascular effects of vasopressin are complex, and vasopressin's role in physiological situations is ill-defined. Vasopressin is a potent vasoconstrictor (V1 receptormediated), and resistance vessels throughout the circulation may be affected (Lszlet al., 1991). Vascular smooth muscle in the skin, skeletal muscle, fat, pancreas, and thyroid gland appear most sensitive, with significant vasoconstriction also occurring in the gastrointestinal tract, coronary vessels, and brain (Liard et al., 1982). However, despite the potency of vasopressin as a direct vasoconstrictor, vasopressin-induced pressor responses in vivo are minimal and occur only with concentrations of vasopressin significantly higher than those required for maximal antidiuresis. To a large extent, this is due to circulating vasopressin that acts on V1 receptors to inhibit sympathetic efferents and potentiate baroreflexes (Abboud et al., 1990). In addition, in some blood vessels, V2 receptors cause vasodilation, perhaps via release of nitric oxide (a potent vasodilator) from the vascular endothelium (Aki et al., 1994), and studies by Manning and coworkers (1999) suggest the existence of a unique vasopressin vasodilator receptor. A large body of data from experimental animals supports the conclusion that vasopressin helps maintain arterial blood pressure during episodes of severe hypovolemia/hypotension (Lszlet al., 1991). However, antagonism of V1 receptors does not alter the hypotensive response to lower-body negative pressure in normal subjects despite a fivefold elevation in circulating vasopressin levels (Hirsch et al., 1993). Vasopressin also may increase total peripheral resistance in heart failure, and administration of a peptide V1 receptor antagonist improves hemodynamic function in such patients (Thibonnier, 1988). At present, there is no convincing evidence for a role of vasopressin in essential hypertension in human beings (Kawano et al., 1997). The effects of vasopressin on the heart (reduced cardiac output and heart rate) are largely indirect and are the result of coronary vasoconstriction, decreased coronary blood flow, and alterations in vagal and sympathetic tone (Lszlet al., 1991). In human beings, the effects of vasopressin on coronary blood flow can be easily demonstrated, especially if large doses are employed. The cardiac actions of the hormone are of more than academic interest. Some patients with coronary insufficiency experience anginal pain even in response to the relatively small amounts of vasopressin required to control diabetes insipidus, and vasopressin-induced myocardial ischemia has led to severe reactions and even death. Central Nervous System It is likely that vasopressin plays a role as a neurotransmitter and/or neuromodulator (Gash et al., 1987; Jolles, 1987). Vasopressin may participate in the acquisition of certain learned behaviors (Dantzer and Bluth, 1993), in the development of some complex social processes (Insel et al., 1993; Young et al., 1998), and in the pathogenesis of specific psychiatric diseases (Legros et al., 1993). However, the physiological/pathophysiological relevance of these findings is controversial, and some of the actions of vasopressin on memory and learned behavior may be due to visceral autonomic effects. In 1931, Cushing reported on the antipyretic effects of pituitary extracts injected into the lateral ventricle of febrile patients. Since then, many studies have supported a physiological role for vasopressin as a naturally occurring antipyretic factor (Kasting, 1989; Cridland and Kasting, 1992). Although vasopressin can modulate CNS autonomic systems controlling heart rate, arterial blood pressure, respiration rate, and sleep patterns, the physiological significance of these actions is unclear. Finally, secretion of ACTH is enhanced by vasopressin that is delivered to the anterior pituitary via a neuronal pathway leading to secretion of peptide into the hypophyseal portal blood. However, vasopressin is not the principal corticotropin-releasing factor. The CNS effects of vasopressin appear to be mediated predominantly by V1 receptors. Blood Coagulation Activation of V2 receptors by desmopressin or vasopressin increases circulating levels of procoagulant factor VIII and of von Willebrand factor (David, 1993). This effect is mediated by extrarenal V2 receptors (Bernat et al., 1997). Presumably, vasopressin stimulates the secretion of von Willebrand factor and of factor VIII from storage sites in vascular endothelium. However, since release of von Willebrand factor does not occur when desmopressin is applied directly to cultured endothelial cells or to isolated blood vessels, intermediate factors are likely to be involved. In this regard, it has been hypothesized that desmopressin releases interleukin 1 (IL-1) from monocytes, and IL-1 can then release von Willebrand factor (Breit and Green, 1988). Other Nonrenal Effects of Vasopressin At high concentrations, vasopressin stimulates uterine (viaoxytocin receptors) and gastrointestinal (via V1 receptors) smooth muscle. Vasopressin is stored in platelets, and V1 receptors stimulate platelet aggregation (Inaba et al., 1988). Also, V1 receptors located on hepatocytes stimulate glycogenolysis (Keppens and de Wulf, 1975). The physiological significance of these effects of vasopressin is not known. |
Diseases Affecting the Vasopressin System
|
Diabetes Insipidus (DI) DI is a disease of impaired renal conservation of water due either to an inadequate secretion of vasopressin from the neurohypophysis (central or cranial DI) or to an insufficient renal response to vasopressin (nephrogenic DI). Very rarely, DI can be caused by an abnormally high rate of degradation of vasopressin by circulating vasopressinases (Durr et al., 1987). Pregnancy may accentuate or reveal central and/or nephrogenic DI by increasing plasma levels of vasopressinase and by reducing the renal sensitivity to vasopressin. Patients with DI excrete large volumes (more than 30 ml/kg per day) of dilute (less than 200 mOsm/kg) urine and, if their thirst mechanism is functioning normally, are polydipsic. In contrast to the sweet urine excreted by patients with diabetes mellitus, urine from patients with DI is tasteless, hence the name, insipidus. Fortunately, the urinary taste test for DI devised by Willis in the seventeenth century has been supplanted by the more palatable approach of simply observing whether the patient is able to reduce urine volume and increase urine osmolality after a period of carefully observed fluid deprivation. Central DI can be distinguished from nephrogenic DI by administration of desmopressin, which will increase urine osmolality in patients with central DI but have little or no effect in patients with nephrogenic DI. DI can be differentiated from primary polydipsia by measuring plasma osmolality, which will be low to low-normal in patients with primary polydipsia and high to high-normal in patients with DI. For a more complete discussion of diagnostic procedures, see Vokes and Robertson (1988). Central DI Head injury, either surgical or traumatic, in the region of the pituitary and/or hypothalamus may cause central DI. Postoperative central DI may be transient, permanent, or triphasic (recovery followed by permanent relapse) (Seckl and Dunger, 1992). Other causes include hypothalamic or pituitary tumors, cerebral aneurysms, CNS ischemia, and brain infiltrations and infections. Finally, central DI may be idiopathic or familial. Familial central DI is usually autosomal dominant (chromosome 20) and has been associated with point mutations in the signal peptide and VP-neurophysin (seeFigure 301), leading to defects in the synthesis, processing, and transport of the preprohormone complex (Raymond, 1994). Since familial central DI is autosomal dominant, the defective preprohormone coded by the mutant allele must interfere in some way with the synthesis of hormone coded by the normal allele. Antidiuretic peptides are the primary treatment for central DI, with desmopressin being the peptide of choice. (See'Clinical Pharmacology of Vasopressin Peptides,' below, for discussion of antidiuretic peptides in the treatment of central DI.) For patients with central DI who cannot tolerate antidiuretic peptides because of side effects or allergic reactions, other treatment options are available. Chlorpropamide, an oral sulfonylurea that potentiates the action of small or residual amounts of circulating vasopressin, will cause a reduction in urine volume in more than half of all patients with central DI at a dose of 125 to 500 mg daily and is particularly effective in patients with partial central DI, where it potentiates the action of low levels of circulating vasopressin. If polyuria is not satisfactorily controlled with chlorpropamide alone, addition of a thiazide diuretic (Chapter 29: Diuretics) to the regimen usually results in an adequate reduction in the volume of urine. Carbamazepine (800 to 1000 mg daily in divided doses) and clofibrate (1 to 2 g daily in divided doses) also reduce urine volume in patients with central DI. Long-term use of these agents may induce serious adverse effects; therefore carbamazepine and clofibrate are rarely used to treat central DI. The antidiuretic mechanism(s) of chlorpropamide, carbamazepine, and clofibrate are not clear. These agents are not effective in nephrogenic DI, which indicates that functional V2 receptors are required for the antidiuretic mechanism to be expressed. Since carbamazepine inhibits and chlorpropamide has little effect on vasopressin secretion, it is likely that carbamazepine and chlorpropamide act directly on the kidney to enhance V2 receptormediated antidiuresis. Nephrogenic DI Nephrogenic DI may be acquired or genetic. Hypercalcemia, hypokalemia, postobstructive renal failure, lithium, and demeclocycline can induce nephrogenic DI. As many as one in three patients treated with lithium may develop nephrogenic DI. Familial, X-linked, recessive nephrogenic DI is caused by mutations in the gene coding for the V2 receptor (a gene located in the q28 region of the X chromosome). A number of missense, nonsense, and frame-shift mutations in the gene encoding the V2 receptor have been identified in patients with familial, X-linked nephrogenic DI (Oksche and Rosenthal, 1998; Morello et al., 2000). Of all mutant alleles reported to date, mutations encoded result predominantly in abnormal synthesis, processing, or intracellular transport of V2 receptors. Autosomal recessive nephrogenic DI is caused by inactivating mutations in aquaporin 2 (13 different mutations have been identified to date). These findings indicate that aquaporin 2 is essential for the antidiuretic effect of vasopressin in human beings (Deen et al., 1994). Although the mainstay of treatment of nephrogenic DI is assurance of an adequate intake of water, drugs also can be used to reduce polyuria. Amiloride (Chapter 29: Diuretics) blocks the uptake of lithium by the sodium channel in the collecting duct system and is therefore the drug of choice for lithium-induced nephrogenic DI. Paradoxically, thiazide diuretics cause a reduction in the polyuria of patients with DI and often are used to treat nephrogenic DI. The use of thiazide diuretics in infants with nephrogenic DI may be crucially important, since uncontrolled polyuria may exceed the child's capacity to imBIBe and absorb fluids. The antidiuretic mechanism of thiazides in DI is incompletely understood. It is possible that the natriuretic action of thiazides and resultant depletion of extracellular fluid volume play an important role in the thiazide-induced antidiuresis in DI. In this regard, whenever extracellular fluid volume is reduced, compensatory mechanisms increase reabsorption of NaCl in the proximal tubule, with a resultant reduction of volume delivered to the distal tubule. Consequently, less free water can be formed, and this should diminish polyuria. However, recent studies in rats with vasopressin-deficient DI seriously challenge this hypothesis (Grnbeck et al., 1998). Nonetheless, the antidiuretic effects appear to parallel the ability of thiazides to cause natriuresis, and the drugs are given in doses similar to those used for mobilization of edema fluid. In patients with DI, a 50% reduction of urine volume is a good response to thiazides. Moderate restriction of sodium intake can enhance the antidiuretic effectiveness of thiazides. A number of case reports describe the effectiveness of indomethacin in the treatment of nephrogenic DI (Libber et al., 1986); however, other prostaglandin synthase inhibitors (e.g., ibuprofen) appear to be less effective. The mechanism of the effects of indomethacin is unclear but may involve a decrease in glomerular filtration rate, an increase in medullary solute concentration, and/or enhanced proximal reabsorption of fluid (Seckl and Dunger, 1992). Also, since prostaglandins attenuate vasopressin-induced antidiuresis in patients with at least a partially intact V2 receptor system, a portion of the antidiuretic response to indomethacin may be due to enhancement of the effects of vasopressin on the principal cells of the collecting duct. Syndrome of Inappropriate Secretion of Antidiuretic Hormone (SIADH) SIADH is a disease of impaired water excretion with accompanying hyponatremia and hypoosmolality caused by the inappropriate secretion of vasopressin. The clinical manifestations of plasma hypotonicity resulting from SIADH may include lethargy, anorexia, nausea/vomiting, muscle cramps, coma, convulsions, and death. A multitude of disorders can induce SIADH (Zerbe et al., 1980) including malignancies, pulmonary diseases, CNS injuries/diseases (head trauma, infections, tumors), general surgery, and drugs (e.g., cisplatin, vinca alkaloids, cyclophosphamide, chloropropamide, thiazide diuretics, phenothiazines, carbamazepine, clofibrate, nicotine, narcotics, and tricyclic antidepressants). In a normal individual, an elevation in plasma vasopressin levels per se does not induce plasma hypotonicity, because the person simply stops drinking due to an osmotically induced aversion to fluids. Therefore, plasma hypotonicity only occurs when excessive fluid intake (oral or intravenous) accompanies inappropriate secretion of vasopressin. Treatment of hypotonicity in the setting of SIADH includes water restriction, intravenous administration of hypertonic saline, loop diuretics (which interfere with the concentrating ability of the kidneys), and drugs that inhibit the ability of vasopressin to increase water permeability in the collecting ducts. To inhibit vasopressin's action in the collecting ducts, demeclocycline is the preferred drug (however, see'Prospectus,' below). Although lithium can inhibit the renal actions of vasopressin, it is effective in only a minority of patients, may induce irreversible renal damage when used chronically, and has a low therapeutic index. Therefore, lithium should be used only in patients with symptomatic SIADH who cannot be controlled by other means or in whom tetracyclines are contraindicated, e.g., patients with liver disease. It is important to stress that the majority of patients with SIADH do not require therapy because plasma Na+ stabilizes in the range of 125 to 132 mM; such patients usually are asymptomatic. Only when symptomatic hypotonicity ensues, generally when plasma Na+ levels drop below 120 mM, should therapy with demeclocycline be initiated. Since hypotonicity, which causes an influx of water into cells with resulting cerebral swelling, is the cause of symptoms, the goal of therapy is simply to increase plasma osmolality toward normal. For a more complete description of the diagnosis and treatment of SIADH, see Kovacs and Robertson (1992). Other Water-Retaining States In patients with congestive heart failure, liver cirrhosis, and nephrotic syndrome, effective blood volume often is reduced, and hypovolemia is frequently exacerbated by the liberal use of diuretics in such patients. Since hypovolemia stimulates vasopressin release, patients may become hyponatremic due to vasopressin-mediated retention of water. The development of potent, orally active V2 receptor antagonists and specific inhibitors of water channels in the collecting duct would provide an effective therapeutic strategy, not only in patients with SIADH but also in the much more common setting of hyponatremia in patients with heart failure, liver cirrhosis, and nephrotic syndrome. |
Clinical Pharmacology of Vasopressin Peptides
|
Therapeutic Uses Only three antidiuretic peptides are available for clinical use in the
V1 receptormediated therapeutic applications are based on the rationale that V1
receptors cause contraction of gastrointestinal and vascular smooth muscle. V1
receptormediated contraction of gastrointestinal smooth muscle is useful to
treat postoperative ileus and abdominal distension and to dispel intestinal
gas before abdominal roentgenography to avoid interfering gas shadows. V1
receptormediated vasoconstriction of the splanchnic arterial vessels reduces
blood flow to the portal system and thereby attenuates pressure and bleeding
in esophageal varices (Burroughs, 1998). Although endoscopic sclero-therapy
is the treatment of choice for bleeding esophageal varices, V1
receptor agonists can be used in an emergency setting until endoscopy can be
performed. Simultaneous administration of nitroglycerin with vasopressin has
been reported to reverse the cardiotoxic effects of vasopressin while
enhancing the beneficial splanchnic effects of the drug (Gimson et al.,
1986). Also, V1 receptor agonists can be used during abdominal
surgery in patients with portal hypertension to diminish the risk of
hemorrhage during the procedure. Finally, V1 receptormediated
vasoconstriction of the gastric vascular bed reduces bleeding in acute
hemorrhagic gastritis (Peterson, 1989). 8-Arginine vasopressin should be used
for all V1 receptormediated therapeutic applications. Although
not yet available in the V2 receptormediated therapeutic applications are based on the rationale that V2
receptors cause water conservation and release of blood coagulation factors.
Central, but not nephrogenic, DI can be treated with V2 receptor
agonists, and polyuria and polydipsia are usually well controlled. Some
patients experience transient DI (e.g., in head injury or surgery in
the area of the pituitary); however, for most patients with DI, therapy is
lifelong. Desmopressin is the drug of choice for the vast majority of
patients, and numerous clinical trials (Robinson, 1976; Cobb et al.,
1978) have demonstrated that desmopressin is an effective agent in both
adults and children and has few side effects. The duration of effect from a
single intranasal dose is from 6 to 20 hours, and twice-daily administration
has proven to be effective in most patients. There is considerable
variability in the intranasal dose of desmopressin required to maintain
normal urine volume, and the dosage must be individually tailored. The usual
intranasal dosage range in adults is 10 to 40 Lypressin nasal spray also can be used to treat central DI; however, lypressin's duration of action is short (4 to 6 hours), making it less convenient than desmopressin. Also, lypressin, like vasopressin, can induce V1 receptormediated adverse effects. Nonetheless, for patients refractory to desmopressin or who experience side effects with desmopressin, lypressin provides an alternative. 8-Arginine vasopressin has little if any place in the long-term therapy of DI because of its short duration of action and V1 receptormediated side effects. Vasopressin can be used as an alternative to desmopressin in the initial diagnostic evaluation of patients with suspected DI and to control polyuria in patients with DI who have recently undergone surgery or experienced head trauma. Under these circumstances, polyuria may be transient, and long-acting agents may produce water intoxication. An additional V2 receptormediated therapeutic application
is the use of desmopressin in bleeding disorders (Mannucci, 1997; Sutor, 1998).
In most patients with type I von Willebrand's disease (vWD) and in some with
type IIN vWD, desmopressin will elevate von Willebrand factor and shorten
bleeding time. However, desmopressin is generally ineffective in patients
with types IIa, IIb, and III vWD. Desmopressin may cause a marked, transient
thrombocytopenia in individuals with type IIb vWD and may be dangerous in
such patients. Desmopressin also increases factor VIII levels in patients
with moderately severe hemophilia A. Desmopressin is not indicated in
patients with severe hemophilia A, those with hemophilia B, or those with
factor VIII antibodies. The response of any given patient with type I vWD or
hemophilia A to desmopressin should be determined at the time of diagnosis or
1 to 2 weeks before elective surgery to assess the extent of increase in
factor VIII or von Willebrand factor. Desmopressin is employed widely to
treat the hemostatic abnormalities induced by uremia (Mannucci et al.,
1983). In patients with renal insufficiency, desmopressin shortens bleeding
time and increases circulating levels of factor VIII coagulant activity,
factor VIIIrelated antigen, and ristocetin cofactor. It also induces the
appearance of larger von Willebrand factor multimers. Desmopressin is
effective in some patients with liver cirrhosisinduced or druginduced (heparin,
hirudin, antiplatelet agents) bleeding disorders. Desmopressin, given
intravenously at a dose of 0.3 Another V2 receptormediated therapeutic application is the use of desmopressin for primary nocturnal enuresis (Sukhai, 1993). In this regard, desmopressin should be used only intranasally and may be used alone or in combination with behavioral conditioning. Finally, desmopressin has been found to relieve postlumbar puncture headache, probably by causing water retention and thereby facilitating rapid fluid equilibration in the CNS. Pharmacokinetics When vasopressin, lypressin, and desmopressin are given orally, they are quickly inactivated by trypsin, which cleaves the peptide bond between amino acids 8 and 9. Inactivation by peptidases in various tissues (particularly the liver and kidneys) results in a plasma half-life of vasopressin of 17 to 35 minutes. The plasma half-life of desmopressin has two components, a fast component of 6.5 to 9 minutes and a slow component of 30 to 117 minutes. Toxicity, Adverse Effects, Contraindications, Drug Interactions Most adverse effects are mediated through the V1 receptor acting on vascular and gastrointestinal smooth muscle; consequently such adverse effects are much less common and less severe with desmopressin than with vasopressin or lypressin. After the injection of large doses of vasopressin, marked facial pallor as a result of cutaneous vasoconstriction commonly is observed. Increased intestinal activity is likely to cause nausea, belching, cramps, and an urge to defecate. Most serious, however, is the effect on the coronary circulation. Vasopressin and lypressin should be administered only at low doses and with extreme caution in individuals suffering from vascular disease, especially disease of the coronary arteries. Other cardiac complications include arrhythmia and decreased cardiac output. Peripheral vasoconstriction and gangrene have been encountered in patients receiving large doses of vasopressin. The major V2 receptormediated adverse effect is water intoxication, which can occur with desmopressin, lypressin, or vasopressin. In this regard, carbamazepine, chlorpropamide, and NSAIDs can potentiate the antidiuretic effects of these peptides. Desmopressin, lypressin, and vasopressin should be used cautiously in disease states in which a rapid increase in extracellular water may impose risks (e.g., in angina, hypertension, heart failure) and should not be used in patients with acute renal failure. Also, it is imperative that these peptides not be administered to patients with primary or psychogenic polydipsia, because severe hypotonic hyponatremia will ensue. Allergic reactions, ranging from urticaria to anaphylaxis, may occur with desmopressin, lypressin, or vasopressin. Intranasal administration may cause local adverse effects in the nasal passages, such as edema, scarring, rhinorrhea, congestion, irritation, pruritus, and ulceration. |
Prospectus
|
It is likely that nonpeptide, orally active V2-selective receptor antagonists will become available in the near future for the treatment of dilutional hyponatremia induced by heart failure, liver cirrhosis, nephrotic syndrome, and SIADH (Schrier et al., 1998; Mayinger and Hensen, 1999). In addition, orally active combined V1a-/V2-selective antagonists may become available for diseases such as congestive heart failure, in which both increased peripheral vascular resistance and dilutional hyponatremia are present (Yatsu et al., 1999). Whether or not sporadic administration of membrane-permeant, nonpeptide V2 receptor antagonists will achieve successful in vivo cell surface-receptor delivery of certain alleles of X-linked nephrogenic DI, as demonstrated in vitro, remains to be ascertained in clinical trials (Morello et al., 2000). Several important new clinical applications of the ancient hormone vasopressin are under active investigation. Vasopressin may prove superior to epinephrine as a pressor agent in cardiopulmonary resuscitation (Chugh et al., 1997), and it may be particularly effective in refractory hypotension after cardiopulmonary bypass (Overland and Teply, 1998). Vasopressin levels in patients with septic shock are inappropriately low, and septic shock patients are extraordinarily sensitive to the pressor actions of vasopressin (Landry et al., 1997). Vasopressin also reverses intractable hypotension in the late phase of hemorrhagic shock (Morales et al., 1999). Thus, vasopressin may become the preferred pressor agent in emergency and critical care medicine. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 7325
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved