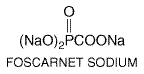| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antimicrobial Agents: Antiviral Agents (Nonretroviral)
Overview
|
The number of antiviral drugs has increased dramatically over the past decade, largely in response to human immunodeficiency virus (HIV) infection and its sequelae (see reviews by Hayden, 2000; Balfour, 1999). This chapter summarizes the agents available for treatment of infections due to DNA and RNA viruses, excluding retroviruses, such as HIV. Many of the available therapeutic agents are directed toward disrupting one of the many steps in viral infection and replication. However, interferons, cytokines that evoke immunomodulating and antiproliferative actions in host cells, also are described (see also Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants). Special sections on antiherpesvirus and antiinfluenza agents are included. The issues related to effective therapy against viruses, including emergence of resistance to particular agents and immunopathological responses to viral antigens, also are discussed. The use of purine and pyrimidine nucleoside analogs for treatment of neoplastic disease, rather than as antiviral agents, is discussed in Chapter 52: Antineoplastic Agents. Antiretroviral agents are discussed in Chapter 51: Antiretroviral Agents: Antiretroviral Agents. |
Antimicrobial Agents: Antiviral Agents (Nonretroviral): Introduction
|
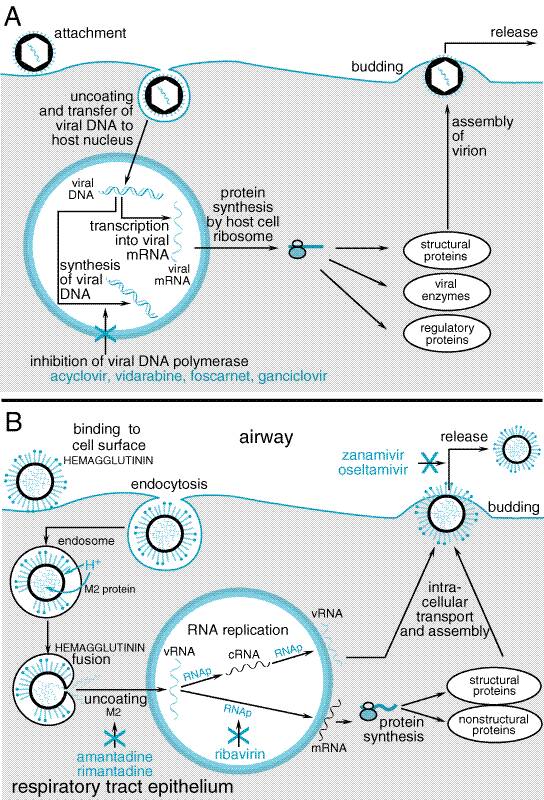
Viruses consist of either double-stranded or single-stranded DNA or RNA enclosed in a protein coat, called a capsid. Some viruses also possess a lipoprotein envelope that, like the capsid, may contain antigenic proteins. Most viruses contain or encode enzymes essential for viral replication inside a host cell. Since viruses have no metabolic machinery of their own, they usurp the machinery of their host cell which, depending on the virus, may be a plant, bacterium, or animal cell. Table 501 outlines the multiple stages of viral replication, which suggest the possibility for development of multiple classes of antiviral agents that could act at each stage of replication. Effective antiviral agents must inhibit virus-specific replicative events or preferentially inhibit virus-directed rather than host celldirected nucleic acid or protein synthesis. The discovery of novel antiviral inhibitors often is linked to a better understanding of the molecular events in viral replication. This chapter provides information about the antiviral activity, pharmacology, and clinical uses of specific antiviral agents for non-HIV infections. Table 502 shows the nomenclature and dosage forms of available antiviral agents. Figure 501 provides a schematic diagram of the replicative cycle of a DNA virus (A) and of an RNA virus (B). DNA viruses (and the diseases they cause) include poxviruses (smallpox), herpesviruses (chickenpox, shingles herpes) adenoviruses (conjunctivitis, sore throat), hepadnaviruses (hepatitis B) and papillomaviruses (warts). Typically, DNA viruses enter into the host cell nucleus, where the viral DNA is transcribed into mRNA by host cell mRNA polymerase; mRNA is translated in the usual host cell fashion into virus-specific proteins. One exception to this strategy is poxvirus, which has its own RNA polymerase and consequently replicates in the host cell cytoplasm.
For RNA viruses, the replication strategy in the host cell relies either on enzymes in the virion (the whole infective viral particle) to synthesize its mRNA or on the viral RNA serving as its own mRNA. The mRNA is translated into various viral proteins, including RNA polymerase, which directs the synthesis of more viral mRNA (see Figure 501B). Certain RNA viruses, such as influenza, have a requirement for active transcription in the host cell nucleus. Examples of RNA viruses (and the diseases they cause) include rubella virus (German measles), rhabdoviruses (rabies), picornaviruses (poliomyelitis, meningitis, colds), arenaviruses (meningitis, Lassa fever), arboviruses (yellow fever, arthropod-borne encephalitis), orthomyxoviruses (influenza), and paramyxoviruses (measles, mumps). One group of RNA viruses that deserve special mention are retroviruses, responsible for diseases such as acquired immunodeficiency syndrome (AIDS; see Chapter 51: Antiretroviral Agents: Antiretroviral Agents) and T-cell leukemias (the human T lymphotropic virus I, HTLV-I). In retroviruses, the virus contains a reverse transcriptase enzyme activity that makes a DNA copy of the viral RNA template. The DNA copy is then integrated into the host genome, at which point it is referred to as a provirus and is transcribed into both genomic RNA and mRNA for translation into viral proteins, giving rise to the generation of new virus particles. Experiences from development of antiviral agents have provided useful general insights that have practical implications. (1) Although many compounds show antiviral activity in vitro, most affect some host cell function and are associated with unacceptable toxicity in human beings. (2) Effective agents typically have a restricted spectrum of antiviral activity and target a specific viral protein, most often an enzyme (polymerase or transcriptase) involved in viral nucleic acid synthesis. (3) Single nucleotide changes leading to critical amino acid substitutions in a target protein often are sufficient to cause antiviral drug resistance. Indeed, the selection of a drug-resistant variant indicates that a drug has a specific antiviral mechanism of action. (4) Current agents inhibit active replication, so that viral growth may resume following drug removal. Effective host immune responses remain essential for recovery from infection. Clinical failures of antiviral therapy may occur with drug-sensitive virus in highly immunocompromised patients or following emergence of drug-resistant variants. Most drug-resistant viruses (e.g., herpesviruses, HIV-1 responsible for AIDS) are recovered from immunocompromised patients with high viral replicative loads and repeated or prolonged courses of antiviral treatment, although influenza A virus is an exception. (5) Current agents do not eliminate nonreplicating or latent virus, although some drugs have been used effectively for chronic suppression of disease reactivation. (6) Clinical efficacy depends on achieving inhibitory concentrations at the site of infection, usually within infected cells. For example, nucleoside analogs must be taken up and phosphorylated intracellularly for activity; consequently, concentrations of critical enzymes or competing substrates influence antiviral effects in cells of different types and metabolic states. (7) In vitro sensitivity tests for antiviral agents are not standardized, and results depend on the assay system, cell type, viral inoculum, and laboratory. Therefore, clear relationships among drug concentrations active in vitro, those achieved in blood or other body fluids, and clinical response have not been established for most antiviral agents. |
Antiherpesvirus Agents
|
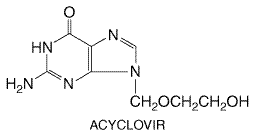
Infection with herpes simplex virus type 1 typically causes diseases of the mouth, face, skin, esophagus, or brain. Herpes simplex virus type 2 usually causes infections of the genitals, rectum, skin, hands, or meninges. In either case, the infection may be a primary one or disease can result from activation of a latent infection. The first systemically administered antiherpesvirus agent of proven value, vidarabine, was approved in 1977. However, its toxicities restricted its use to life-threatening herpes simplex virus (HSV) and varicella zoster virus (VZV) infections. The discovery and development of acyclovir, initially approved in 1982, provided the first effective treatment for less severe HSV and VZV infections in ambulatory patients. Subsequent trials also found that intravenous acyclovir is superior to vidarabine in regard to efficacy and/or toxicity in HSV encephalitis and in VZV infections of immunocompromised patients. Acyclovir is the prototype of a group of antiviral agents that are phosphorylated intracellularly by a viral kinase to become inhibitors of viral DNA synthesis. Other agents employing this strategy include penciclovir and ganciclovir. Acyclovir and Valacyclovir Chemistry and Antiviral Activity Acyclovir (9-[(2-hydroxy-ethoxy)methyl]-9H-guanine) is an acyclic
guanine nucleoside analog that lacks a 3'-hydroxyl on the side
chain. Acyclovir is available as capsules, as an ointment, and as a powder to
be reconstituted for intravenous use. Valacyclovir is the L-valyl ester prodrug of acyclovir.
Acyclovir's clinically useful antiviral spectrum is limited to herpesviruses.
In vitro it is most active against HSV-1 (0.02 to 0.9
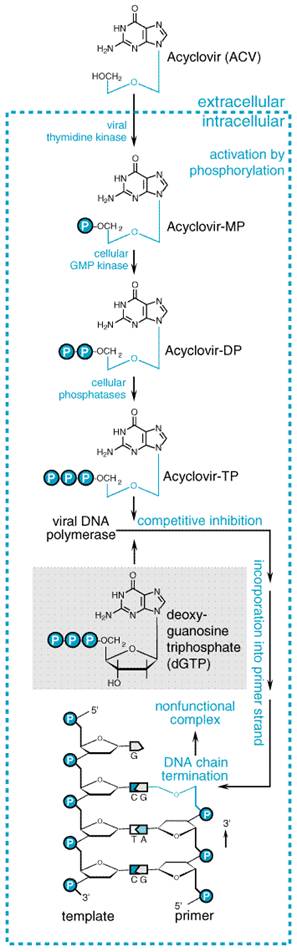
Mechanisms of Action and Resistance Acyclovir inhibits viral DNA synthesis via a mechanism outlined in Figure 502 (Elion, 1986). Its selectivity of action depends on interaction with two distinct viral proteins. Cellular uptake and initial phosphorylation are facilitated by HSV thymidine kinase. The affinity of acyclovir for HSV thymidine kinase is about 200-fold greater than for the mammalian enzyme. Cellular enzymes convert the monophosphate to acyclovir triphosphate, which is present in 40- to 100-fold higher concentrations in HSV-infected than in uninfected cells, and competes for endogenous deoxyguanosine triphosphate (dGTP). The immunosuppressive agent mycophenolate mofetil (see Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants) potentiates the antiherpes activity of acyclovir and related agents by depleting intracellular dGTP pools (Neyts et al., 1998). Acyclovir triphosphate competitively inhibits viral DNA polymerases and, to a much smaller extent, cellular DNA polymerases. Acyclovir triphosphate also is incorporated into viral DNA, where it acts as a chain terminator because of the lack of 3'-hydroxyl group. By a mechanism termed suicide inactivation, the terminated DNA template containing acyclovir binds the enzyme and leads to irreversible inactivation of the DNA polymerase.
Acyclovir
resistance in HSV has been linked to one of three mechanisms: absence or
partial production of viral thymidine kinase, altered thymidine kinase
substrate specificity (e.g., phosphorylation of thymidine but not
acyclovir), or altered viral DNA polymerase. Alterations in viral enzymes are
caused by point mutations or base insertions or deletions in the
corresponding genes. Resistant variants are present in native virus
populations, and heterogeneous mixtures of viruses occur in isolates from
treated patients. The most common resistance mechanism in clinical HSV
isolates is deficient thymidine kinase activity (Hill et al., 1991).
Less common is altered thymidine kinase activity; DNA polymerase mutants are
rare. Phenotypic resistance typically is defined by in vitro
inhibitory concentrations >2 to 3 Acyclovir resistance in VZV isolates is caused by mutations in VZV thymidine kinase or less often by mutations in viral DNA polymerase. Absorption, Distribution, and Elimination Table 503 compares the pharmacokinetic properties of acyclovir with
those of other antiherpesvirus agents. The oral bioavailability of acyclovir
ranges from 10% to 30% and decreases with increasing dose (Wagstaff et al.,
1994). Peak plasma concentrations average 0.4 to 0.8 Valacyclovir is converted rapidly and virtually completely to acyclovir
after oral administration in healthy adults. This conversion is thought to
result from first-pass intestinal and hepatic metabolism through enzymatic
hydrolysis. Unlike acyclovir, valacyclovir is a substrate for intestinal and
renal peptide transporters (Ganapathy et al., 1998). The relative oral
bioavailability of acyclovir increases three- to fivefold to approximately
70% following valacyclovir administration (Steingrimsdottir et al.,
2000). Peak acyclovir concentrations average 5 to 6 Acyclovir distributes widely in body fluids including vesicular fluid, aqueous humor, and cerebrospinal fluid. Compared to plasma, salivary concentrations are low, and vaginal secretion concentrations vary widely. Acyclovir is concentrated in breast milk, amniotic fluid, and placenta. Newborn plasma levels are similar to maternal ones (Frenkel et al., 1991). Percutaneous absorption of acyclovir after topical administration is low. The mean plasma half-life (t1/2) of elimination of acyclovir is about 2.5 hours, with a range of 1.5 to 6 hours in adults with normal renal function. The plasma t1/2 of elimination of acyclovir is about 4 hours in neonates and increases to 20 hours in anuric patients (Blum et al., 1982). Renal excretion of unmetabolized acyclovir by glomerular filtration and tubular secretion is the principal route of elimination. Less than 15% is excreted as 9-carboxymethoxymethylguanine or minor metabolites. The pharmacokinetics of oral acyclovir and valacyclovir appear to be similar in pregnant and nonpregnant women (Kimberlin et al., 1998). Untoward Effects Acyclovir generally is well tolerated. Topical acyclovir in a polyethylene glycol base may cause mucosal irritation and transient burning when applied to genital lesions. Oral acyclovir has been associated infrequently with nausea, diarrhea, rash, or headache and very rarely with renal insufficiency or neurotoxicity. Valacyclovir also may be associated with headache, nausea, and diarrhea. Chronic acyclovir suppression of genital herpes has been used safely for over 5 years (Goldberg et al., 1993). No excess frequency of abnormalities has been recognized in infants born to women exposed to acyclovir during pregnancy (Reiff-Eldridge et al., 2000). The tolerance profile of valacyclovir appears to be similar to that of oral acyclovir. High doses of valacyclovir have been associated with confusion, hallucinosis, nephrotoxicity and, uncommonly, with severe thrombocytopenic syndromes, sometimes fatal, in immunocompromised patients (Feinberg et al., 1998). The principal dose-limiting toxicities of intravenous acyclovir are
renal insufficiency and central nervous system side effects. Preexisting
renal insufficiency, high doses, and high acyclovir plasma levels (>25 Severe somnolence and lethargy may occur with combinations of zidovudine and acyclovir. Concomitant cyclosporine and probably other nephrotoxic agents enhance the risk of nephrotoxicity. Probenecid decreases the renal clearance and prolongs the plasma t1/2 of elimination. Acyclovir may decrease the renal clearance of other drugs eliminated by active renal secretion, such as methotrexate. Therapeutic Uses In immunocompetent persons, the clinical benefits of acyclovir are greater in initial HSV infections than in recurrent ones, which typically are milder in severity (Whitley and Gnann, 1992). Acyclovir is particularly useful in immunocompromised patients, because these individuals experience both more frequent and more severe HSV and VZV infections. Since VZV is less susceptible than HSV to acyclovir, higher doses must be used for treating varicella or zoster cases than for HSV infections. Oral valacyclovir is as effective as oral acyclovir in HSV infections and more effective for treating herpes zoster. Herpes Simplex Virus Infections In initial genital HSV infections, oral acyclovir (200 mg five times daily for 10 days) and valacyclovir (1000 mg twice daily for 10 days) are associated with significant reductions in virus shedding, symptoms, and time to healing (Fife et al., 1997). Intravenous acyclovir (5 mg/kg per 8 hours) has similar effects in patients hospitalized with severe primary genital HSV infections. Topical acyclovir is much less effective than systemic administration. None of these regimens reproducibly reduces the risk of recurrent genital lesions. Patient-initiated acyclovir (200 mg 5 times daily for 5 days) or valacyclovir (500 mg or 1000 mg twice daily for 5 days) shortens the manifestations of recurrent genital HSV episodes by 1 to 2 days (Tyring et al., 1998b). Topical acyclovir offers no significant clinical benefit in recurrent genital herpes. Frequently recurring genital herpes can be suppressed effectively with chronic oral acyclovir (400 mg two times daily or 200 mg three times daily) (Goldberg et al., 1993) or with valacyclovir (500 or 1000 mg once daily) (Patel et al., 1997). During use, recurrences decrease by about 90%, and the majority of patients are free from symptomatic recurrences for periods up to 5 years. Asymptomatic shedding may occur during suppression, as may HSV transmission to sexual partners. Chronic suppression may be useful in those with disabling recurrences of herpetic whitlow or HSV-related erythema multiforme. Oral acyclovir is effective in primary herpetic gingivostomatitis (600
mg/m2 four times daily for 10 days in children) but provides
modest clinical benefit in recurrent orolabial herpes. Topical acyclovir
ointment is not clinically beneficial in recurrent herpes labialis. Topical
acyclovir cream, not available in the In immunocompromised patients with mucocutaneous HSV infection, intravenous acyclovir (250 mg/m2 per 8 hours for 7 days) shortens healing time, duration of pain, and the period of virus shedding (Wade et al., 1982). Oral acyclovir (800 mg five times per day) also is effective. Recurrences are common after cessation of therapy and may require long-term suppression. In those with very localized labial or facial HSV infections, topical acyclovir may provide some benefit. Intravenous acyclovir may be beneficial in viscerally disseminating HSV in immunocompromised patients and in HSV-infected burn wounds. Systemic acyclovir prophylaxis is highly effective in preventing mucocutaneous HSV infections in seropositive patients undergoing immunosuppression. Intravenous acyclovir (250 mg/m2 every 8 to 12 hours), begun prior to transplantation and continuing for several weeks, prevents HSV disease in bone-marrow transplant recipients. For patients who can tolerate oral medications, oral acyclovir (400 mg five times/day) is effective, and long-term oral acyclovir (200 to 400 mg three times a day for 6 months) also reduces the risk of VZV infection (Steer et al., 2000). Oral acyclovir prophylaxis also is effective in transplant patients and in those on chemotherapy. In HSV encephalitis, acyclovir (10 mg/kg per 8 hours for a minimum of 10 days) reduces mortality by over 50% and improves overall neurologic outcome compared to vidarabine (Whitley et al., 1986). Higher doses (15 to 20 mg/kg per 8 hours and treatment to 21 days) are recommended by some experts. Intravenous acyclovir (20 mg/kg per 8 hours for 21 days) is more effective than lower doses in neonatal HSV infections (Kimberlin et al., 1999). In neonates and immunosuppressed patients, and, rarely, in previously healthy persons, relapses of encephalitis following acyclovir indicate that longer courses of treatment are needed. An ophthalmic formulation of acyclovir, not available in the In immunocompromised hosts, acyclovir-resistant HSV isolates can cause extensive mucocutaneous disease and rarely meningoencephalitis, pneumonitis, or visceral disease. Infection due to resistant HSV is rare in immunocompetent persons. Resistant HSV can be recovered from 6% to 17% of immunocompromised patients receiving acyclovir treatment (Christophers et al., 1998; Englund et al., 1990). Recurrences after cessation of acyclovir usually are due to sensitive virus but may be due to acyclovir-resistant virus in AIDS patients. Limited acyclovir-resistant HSV infections sometimes undergo spontaneous healing after acyclovir treatment is terminated. In patients with progressive disease, intravenous foscarnet therapy is effective, but vidarabine is not (Safrin et al., 1991). Varicella Zoster Virus Infections If begun within 24 hours of rash onset, oral acyclovir has therapeutic effects in varicella infections of children and adults. In children up to 40 kg body weight, acyclovir (20 mg/kg, up to 800 mg per dose, four times daily for 5 days) reduces fever and new lesion formation by about 1 day. Routine use in uncomplicated pediatric varicella is not recommended, but should be considered in those at risk of moderate to severe illness (persons over 12 years old, secondary household cases, those with chronic cutaneous or pulmonary disorders, or those receiving corticosteroids or long-term salicylates) (Committee on Infectious Diseases, 2000). In adults, early oral acyclovir (800 mg five times daily for 7 days) reduces the time to crusting of lesions by approximately 2 days, the maximum number of lesions by one-half, and the duration of fever (Wallace et al., 1992). Later treatment is not beneficial. Intravenous acyclovir appears to be effective in varicella pneumonia or encephalitis of previously healthy adults. Oral acyclovir (10 mg/kg 4 times daily) given between 7 through 14 days after exposure appears to be effective prophylaxis for varicella (Kumagai et al., 1999). In older adults with localized herpes zoster, oral acyclovir (800 mg
five times daily for 7 days) reduces pain and healing times if treatment can
be initiated within 72 hours of rash onset (Wood et al., 1998). A
reduction in ocular complications, particularly keratitis and anterior
uveitis, occurs with treatment of zoster ophthalmicus (Cobo et al.,
1986). Prolonged acyclovir and concurrent prednisone for 21 days speed zoster
healing and improve quality of life measures compared to each therapy alone (Whitley
et al., 1996). Valacyclovir (1000 mg three times daily for 7 days)
provides more prompt relief of zoster-associated pain than acyclovir in acute
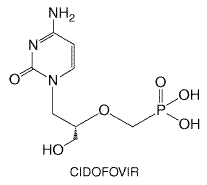
herpes zoster of older adults ( In immunocompromised patients with herpes zoster, intravenous acyclovir (500 mg/m2 per 8 hours for 7 days) reduces viral shedding, healing times, the risks of cutaneous dissemination and visceral complications, as well as the length of hospitalization in disseminating zoster. In immunosuppressed children with varicella, intravenous acyclovir decreases healing times and the risk of visceral complications. Acyclovir-resistant VZV isolates uncommonly have been recovered from HIV-infected children and adults who may manifest chronic hyperkeratotic or verrucous lesions. Meningoradiculitis due to resistant virus also has been described. Intravenous foscarnet also appears to be effective for acyclovir-resistant VZV infections. Other Viruses Acyclovir is ineffective therapeutically in established cytomegalovirus (CMV) infections but has been used for CMV prophylaxis in immunocompromised patients. High-dose intravenous acyclovir (500 mg/m2 per 8 hours for 1 month) in CMV-seropositive bone-marrow transplant recipients is associated with about 50% lower risk of CMV disease and, when combined with prolonged oral acyclovir (800 mg four times daily through 6 months), improves survival (Prentice et al., 1994). High-dose oral acyclovir suppression for 3 months may reduce the risk of CMV disease in certain solid-organ transplant recipients. In seronegative renal transplant patients receiving seropositive donations, valacyclovir (2000 mg 4 times daily for 90 days) prophylaxis reduces CMV disease, other infections, and acute graft rejection risk (Lowance et al., 1999). Compared to acyclovir, high-dose valacyclovir reduces CMV disease in advanced HIV infection but is associated with greater toxicity and possibly shorter survival (Feinberg et al., 1998). In infectious mononucleosis, acyclovir is associated with transient antiviral effects but no clinical benefits. Epstein-Barr virus (EBV)related oral hairy leukoplakia may improve with acyclovir. Cidofovir Chemistry and Antiviral Activity Cidofovir
(1-[(S)-3-hydroxy-2-(phosphonomethoxy)-propyl]cytosine dihydrate) is a
cytidine nucleotide analog with inhibitory activity against human herpes,
papilloma, polyoma, pox, and adenoviruses (Hitchcock et al., 1996). In
vitro inhibitory concentrations range from <0.2 to 0.7
Mechanisms of Action and Resistance Cidofovir inhibits viral DNA synthesis by slowing and eventually
terminating chain elongation. Cidofovir is metabolized to its active
diphosphate form by cellular enzymes; the levels of phosphorylated
metabolites are similar in infected and uninfected cells. The diphosphate
acts as both a competitive inhibitor with respect to dCTP and as an
alternative substrate for viral DNA polymerase. The diphosphate has a
prolonged intracellular half-life and competitively inhibits CMV and HSV DNA
polymerases at concentrations 8- to 600-fold lower than those required to
inhibit human DNA polymerases (Hitchcock et al., 1996). A
phosphocholine metabolite has a prolonged intracellular half-life ( Cidofovir resistance in CMV is due to mutations in viral DNA polymerase. Low-level resistance to cidofovir develops in a minority of patients by 3 months of therapy (Jabs et al., 1998). Highly ganciclovir-resistant CMV isolates that possess DNA polymerase and UL97 kinase mutations are resistant to cidofovir, and prior ganciclovir therapy may select for cidofovir resistance. Some foscarnet-resistant CMV isolates show cross-resistance to cidofovir, and triple-drug resistant variants with DNA polymerase mutations occur (Tatarowicz et al., 1992). Absorption, Distribution, and Elimination Cidofovir is dianionic at physiological pH and has very low oral
bioavailability (Cundy, 1999). The plasma levels after intravenous dosing
decline in a biphasic pattern with a terminal half-life that averages about
2.6 hours (Cundy et al., 1995b). The volume of distribution
approximates total body water. Penetration into the central nervous system
(CNS) or eye have not been well characterized; low cerebrospinal fluid (CSF)
levels were found in one patient with progressive multifocal
leukoencephalopathy. Topical cidofovir gel may result in low plasma concentrations
(<0.5 Cidofovir is cleared by the kidney via glomerular filtration
and tubular secretion. Over 90% of the dose is recovered unchanged in the
urine without significant metabolism in human beings. The probenecid-sensitive
organic anion transporter 1 mediates uptake of cidofovir into proximal renal
tubular epithelial cells (Ho et al., 2000). High-dose probenecid (2 g
3 hours before and 1 g 2 and 8 hours after each infusion) blocks tubular
transport of cidofovir and reduces renal clearance and associated
nephrotoxicity. At cidofovir doses of 5 mg/kg, peak plasma concentrations
increase from 11.5 to 19.6 Untoward Effects Nephrotoxicity is the principal dose-limiting side effect of intravenous cidofovir. Proximal tubular dysfunction includes proteinuria, azotemia, glycosuria, metabolic acidosis, and uncommonly Fanconi's syndrome. Concomitant oral probenecid (see above) and saline prehydration reduce the risk of renal toxicity. On maintenance doses of 5 mg/kg every two weeks, up to 50% of patients develop proteinuria, 10% to 15% elevated serum creatinine, and 15% to 20% neutropenia. Anterior uveitis, responsive to topical corticosteroids and cycloplegia, occurs commonly and ocular hypotony infrequently with intravenous cidofovir (Ambati et al., 1999). Concurrent probenecid is associated with gastrointestinal upset, constitutional symptoms, and hypersensitivity reactions including fever, rash, and, uncommonly, anaphylactoid manifestations. Administration with food and pretreatment with antiemetics, antihistamines, and/or acetaminophen may improve tolerance. Probenecid but not cidofovir alters zidovudine pharmacokinetics, such
that zidovudine doses should be reduced on probenecid-administration days.
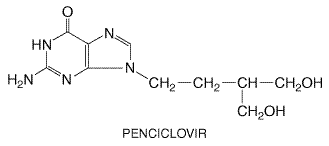
The excretion of other agents affected by probenecid [e.g., Topical application of cidofovir is associated with dose-related application-site reactions (burning, pain, pruritus) in up to one-third of patients and occasionally ulceration (Lalezari et al., 1997). Intravitreal cidofovir may cause vitreitis, hypotony, and visual loss and is contraindicated. Preclinical studies indicate that cidofovir has mutagenic, gonadotoxic, embryotoxic, and teratogenic effects. Because cidofovir is carcinogenic in rats, although not in monkeys, this agent is considered a potential human carcinogen. It may cause infertility and is contraindicated during pregnancy. Therapeutic Uses Intravenous cidofovir is approved for the treatment of CMV retinitis in HIV-infected patients. Intravenous cidofovir (5 mg/kg once a week for 2 weeks followed by dosing every 2 weeks) increases the time to progression of CMV retinitis in previously untreated patients and in those failing or intolerant of ganciclovir and foscarnet therapy (Lalezari et al., 1998; Safrin et al., 1997). CMV viremia may persist during cidofovir administration. Maintenance doses of 5 mg/kg are more effective but less well tolerated than 3 mg/kg doses (Anonymous, 1997). Intravenous cidofovir has been used for treating acyclovir-resistant mucocutaneous HSV infection (Lalezari et al., 1994), adenovirus disease in transplant recipients, and progressive multifocal leukoencephalopathy and extensive molluscum contagiosum in HIV patients. Topical cidofovir gel eliminates virus shedding and lesions in some HIV-infected patients with acyclovir-resistant mucocutaneous HSV infections (Lalezari et al., 1997) and has been used in treating anogenital warts and molluscum contagiosum in immunocompromised patients and cervical intraepithelial neoplasia in women. Intralesional cidofovir induces remissions in adults or children with respiratory papillomatosis (Snoeck et al., 1998). An ophthalmic formulation is under study in adenoviral keratoconjunctivitis. Docosanol Docosanol is a long-chain saturated alcohol that has been approved by the United States Food and Drug Administration (FDA) as a 10% over-the-counter cream for treatment of recurrent orolabial herpes. Doconsanol inhibits the in vitro replication of many lipid enveloped viruses, including HSV, at millimolar concentrations. It does not directly inactivate HSV but appears to block fusion between the cellular and viral envelope membranes and inhibit viral entry into the cell (Pope et al., 1998). Topical treatment beginning within 12 hours of prodromal symptoms or lesion onset reduces healing time by about one day and appears to be well tolerated (Anonymous, Medical Letter, 2000). Treatment initiation at papular or later stages provides no benefit. Famciclovir and Penciclovir Chemistry and Antiviral Activity Famciclovir is the diacetyl ester prodrug of 6-deoxy penciclovir and lacks intrinsic antiviral activity. Penciclovir (9-[4-hydroxy-3-hydroxymethylbut-1-yl] guanine) is an acyclic guanine nucleoside analog. Its structure is given below:
Penciclovir is similar to acyclovir in its spectrum of activity and potency against HSV and VZV (Boyd et al., 1993). The side chain differs structurally in that the oxygen has been replaced by a carbon and an additional hydroxymethyl group is present. The inhibitory concentrations of penciclovir depend on cell type but are usually within twofold of those of acyclovir for HSV and VZV (Boyd et al., 1993). It also is inhibitory for hepatitis B virus (HBV). Mechanisms of Action and Resistance Penciclovir is an inhibitor of viral DNA synthesis. In HSV- or VZV-infected cells, penciclovir is initially phosphorylated by viral thymidine kinase. Penciclovir triphosphate serves as a competitive inhibitor of viral DNA polymerase (Vere Hodge, 1993; see also Figure 502). Although penciclovir triphosphate is approximately 100-fold less potent in inhibiting viral DNA polymerase than is acyclovir triphosphate, it is present in much higher concentrations and for more prolonged periods in infected cells than acyclovir triphosphate. The prolonged intracellular t1/2 of penciclovir triphosphate, which ranges from 7 to 20 hours, is associated with prolonged antiviral effects. Because it has a 3'-hydroxyl group, penciclovir is not an obligate chain terminator, but it does inhibit DNA elongation. Resistant variants due to thymidine kinase or DNA polymerase mutations can be selected by passage in vitro, but the occurrence of resistance during clinical use is currently low. Thymidine kinasedeficient, acyclovir-resistant herpes viruses are cross-resistant to penciclovir. Absorption, Distribution, and Elimination Oral penciclovir has low (5%) bioavailability. In contrast, famciclovir
is well absorbed orally and rapidly converted to penciclovir by deacetylation
of the side chain and oxidation of the purine ring during and following
absorption from the intestine (Gill and Wood, 1996). Although poorly absorbed
itself, the bioavailability of penciclovir is 65% to 77% following oral
administration of famciclovir. Food slows absorption but does not reduce
overall bioavailability. After single 250- or 500-mg doses of famciclovir,
the peak plasma concentration of penciclovir averages 1.6 and 3.3 Untoward Effects Oral famciclovir is well tolerated but may be associated with
headache, diarrhea, and nausea (Saltzman et al., 1994). Its short-term
tolerance is comparable to that of acyclovir. Urticaria, rash, and,
predominantly in the elderly, hallucinations or confusional states have been
reported. Topical penciclovir, which is formulated in 40% propylene glycol
and a cetomacrogol base, is associated with application-site reactions at low
rates ( Penciclovir is mutagenic at high concentrations in vitro. Studies in laboratory animals indicate that chronic famciclovir administration is tumorigenic and decreases spermatogenesis and fertility in rodents and dogs, but long-term administration (1 year) does not affect spermatogenesis in men (Sacks et al., 1998). No teratogenic effects have been observed in animals, but safety during pregnancy has not been established. No clinically important drug interactions have been identified to date with famciclovir or penciclovir (Gill and Wood, 1996). Therapeutic Uses Oral famciclovir, topical penciclovir, and intravenous penciclovir are approved for managing HSV and VZV infections in various countries (Sacks and Wilson, 1999). Oral famciclovir (250 mg three times a day for 5 to 10 days) is as effective as acyclovir in treating first-episode genital herpes (Loveless et al., 1997). In patients with recurrent genital HSV, patient-initiated famciclovir treatment (125 or 250 mg twice a day for 5 days) reduces healing time and symptoms by about 1 day. Famciclovir (250 mg twice a day for up to one year) is effective for suppression of recurrent genital HSV, but single daily doses are less effective (Diaz-Mitoma et al., 1998). Higher doses (500 mg twice a day) reduce HSV recurrences in HIV-infected persons. Intravenous penciclovir (5 mg/kg per 8 or 12 hours for 7 days) is comparable to intravenous acyclovir for treating mucocutaneous HSV infections in immunocompromised hosts (Lazarus et al., 1999). In immunocompetent persons with recurrent orolabial HSV, topical 1% penciclovir cream (applied every 2 hours while awake for 4 days) shortens healing time and symptoms by about 1 day (Spruance et al., 1997). In immunocompetent adults with herpes zoster of 3 days duration or
less, famciclovir (500 mg three times a day for 10 days) is at least as
effective as acyclovir (800 mg 5 times daily) in reducing healing time and
zoster-associated pain, particularly in those aged Famciclovir is associated with dose-related reductions in hepatitis B virus (HBV) DNA and transaminase levels in patients with chronic HBV hepatitis (Trepo et al., 2000) and has been used for recurrent HBV infection following liver transplantation. However, it appears to be less potent than lamivudine and is ineffective in treating lamivudine-resistant HBV infections due to emergence of multiply resistant variants (Mutimer et al., 2000). Fomivirsen Fomivirsen, a 21-mer phosphorothioate oligionucleotide, is the first FDA-approved antisense therapy for viral infections. It is complementary to the messenger RNA sequence for the major immediate-early transcriptional region of CMV and inhibits CMV replication through sequence-specific and nonspecific mechanisms, including inhibition of virus binding to cells (Anderson et al., 1996). Fomivirsen is active against CMV strains resistant to ganciclovir, foscarnet, and cidofovir. CMV variants with 10-fold reduced susceptibility to fomivirsen have been selected by in vitro passage (Mulamba et al., 1998). Fomivirsen is given by intravitreal injection in the treatment of CMV
retinitis for patients intolerant of or unresponsive to other therapies (Perry
and Balfour, 1999). In monkeys, the half-life from the vitreous is about 24
hours and from the retina is up to 78 hours (Leeds et al., 1998).
Local metabolism by exonucleases accounts for elimination. In HIV-infected
patients with refractory, sight-threatening CMV retinitis, fomivirsen
injections (330 Foscarnet Chemistry and Antiviral Activity Foscarnet (trisodium phosphonoformate) is an inorganic pyrophosphate analog that is inhibitory for all herpesviruses and HIV (Oberg, 1989; Wagstaff and Bryson, 1994). Its structure is given below:
In vitro
inhibitory concentrations are generally 100 to 300 Mechanisms of Action and Resistance Foscarnet inhibits viral nucleic acid synthesis by interacting
directly with herpesvirus DNA polymerase or HIV reverse transcriptase (Oberg,
1989; Chrisp and Clissold, 1991; see Figure 501B). It is taken
up slowly by cells and does not undergo significant intracellular metabolism.
Foscarnet reversibly blocks the pyrophosphate binding site of the viral
polymerase in a noncompetitive manner and inhibits cleavage of pyrophosphate
from deoxynucleotide triphosphates. Foscarnet has approximately 100-fold
greater inhibitory effects against herpesvirus DNA polymerases than against
cellular DNA polymerase Herpesviruses resistant to foscarnet have point mutations in the viral DNA polymerase and are associated with three- to sevenfold reductions in vitro (Safrin et al., 1994; Schmit and Boivin, 1999). Absorption, Distribution, and Elimination Oral bioavailability of foscarnet is low (see Table 503).
Following an intravenous infusion of 60 mg/kg per 8 hours, peak and trough
plasma concentrations are approximately 450 to 575 Over 80% of foscarnet is excreted unchanged in the urine by glomerular
filtration and probably tubular secretion. Plasma clearance decreases proportionately
with creatinine clearance, and dose adjustments are indicated for small
decreases in renal function. Plasma elimination is complex, with initial
bimodal half-lives totaling 4 to 8 hours and a prolonged terminal t1/2
for elimination averaging 3 to 4 days. Sequestration in bone with gradual
release accounts for the fate of an estimated 10% to 20% of a given dose.
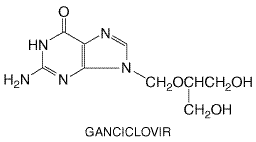
Foscarnet is cleared efficiently by hemodialysis ( Untoward Effects Foscarnet's major dose-limiting toxicities are nephrotoxicity and symptomatic hypocalcemia. Increases in serum creatinine occur in up to one-half of patients but are reversible after cessation in most patients. High doses, rapid infusion, dehydration, prior renal insufficiency, and concurrent nephrotoxic drugs are risk factors. Acute tubular necrosis, crystalline glomerulopathy, nephrogenic diabetes insipidus, and interstitial nephritis have been described. Saline loading may reduce the risk of nephrotoxicity. Foscarnet is highly ionized at physiologic pH, and metabolic abnormalities are very common. These include increases or decreases in Ca2+ and phosphate, hypomagnesemia, and hypokalemia. Decreased serum ionized Ca2+ may cause paresthesia, arrhythmias, tetany, seizures, and other central nervous system disturbances. Concomitant intravenous pentamidine administration increases the risk of symptomatic hypocalcemia. Parenteral magnesium sulfate does not alter foscarnet-induced hypocalcemia or symptoms (Huycke et al., 2000). CNS side effects include headache in about one-fourth of patients, tremor, irritability, seizures, and hallucinosis. Other reported side effects are generalized rash, fever, nausea or emesis, anemia, leukopenia, abnormal liver function tests, electrocardio-graphic (EKG) changes, infusion-related thrombophlebitis, and painful genital ulcerations. Topical foscarnet may cause local irritation and ulceration, and oral foscarnet may cause gastrointestinal disturbance. Preclinical studies indicate that high foscarnet concentrations are mutagenic and that it may cause tooth and skeletal abnormalities in developing laboratory animals. Safety in pregnancy or childhood is uncertain. Therapeutic Uses Intravenous foscarnet is effective for treatment of CMV retinitis, including ganciclovir-resistant infections, and of acyclovir-resistant HSV and VZV infections. It also is effective for treating other types of CMV infections (Wagstaff and Bryson, 1994). Foscarnet is poorly soluble in aqueous solutions and requires large volumes for administration. In CMV retinitis in AIDS patients foscarnet (60 mg/kg per 8 hours or 90 mg/kg per 12 hours for 14 to 21 days followed by chronic maintenance at 90 to 120 mg/kg per day in one dose) is associated with clinical stabilization in about 90% of patients (Wagstaff and Bryson, 1994). A comparative trial of foscarnet with ganciclovir found comparable control of CMV retinitis in AIDS patients but improved overall survival in the foscarnet-treated group (Studies of Ocular Complications of AIDS Research Group, 1992). This improved survival with foscarnet may be related to foscarnet's intrinsic anti-HIV activity (Bergdahl et al., 1998), but patients stop taking foscarnet over three times as often as ganciclovir because of side effects. A combination of foscarnet and ganciclovir is more effective than either drug alone in refractory retinitis (Anonymous, 1996). Foscarnet benefits other CMV syndromes in AIDS or transplant patients but is ineffective as a single drug in treating CMV pneumonia in bone-marrow transplant patients (Oberg, 1989). When used for preemptive therapy of CMV antigenemia in bone-marrow transplant recipients, foscarnet (90 mg/kg per 12 hours for 15 days) is at least as effective as intravenous ganciclovir (Moretti et al., 1998). When used for CMV infections, foscarnet may reduce the risk of Kaposi's sarcoma in HIV-infected patients (Glesby et al., 1996). Intravitreal injections of foscarnet have been used. In acyclovir-resistant mucocutaneous HSV infections, lower doses of foscarnet (40 mg/kg per 8 hours for 7 days or longer) are associated with cessation of viral shedding and with complete healing of lesions in about three-quarters of patients (Safrin et al., 1991). Foscarnet also appears to be effective in acyclovir-resistant VZV infections. Topical foscarnet cream is ineffective in treating recurrent genital HSV in immunocompetent persons but appears to be useful in chronic, acyclovir-resistant infections in immunocompromised patients (Javaly et al., 1999). Resistant clinical isolates of herpesviruses have emerged during therapeutic use (Birch et al., 1992; Safrin et al., 1994) and may be associated with poor clinical response to foscarnet treatment. Ganciclovir and Valganciclovir Chemistry and Antiviral Activity Ganciclovir (9-[1,3-dihydroxy-2-propoxymethyl] guanine) is an acyclic guanine nucleoside analog, similar in structure to acyclovir except in having an additional hydroxymethyl group on the acyclic side chain. Valganciclovir (CYMEVAL) is the L-valyl ester prodrug of ganciclovir. The structure of ganciclovir is given below:
This agent has inhibitory activity against all herpesviruses but is
especially active against CMV (Noble and Faulds, 1998). Inhibitory
concentrations are similar to those of acyclovir for HSV and VZV but 10- to
100-fold lower for human CMV strains (0.2 to 2.8 Inhibitory concentrations for human bone marrow progenitor cells are
similar to those inhibitory for CMV replication, a finding predictive of ganciclovir's
myelotoxicity during clinical use. Inhibition of human lymphocyte blastogenic
responses also occurs at clinically achievable concentrations of 1 to 10 Mechanisms of Action and Resistance Ganciclovir inhibits viral DNA synthesis. It is monophosphorylated intracellularly by a virus-induced enzyme. Phosphorylation is catalyzed by a viral thymidine kinase during HSV infection and by a viral phosphotransferase encoded by the UL97 gene during CMV infection. Ganciclovir di- and triphosphate are formed by cellular enzymes. At least 10-fold higher concentrations of ganciclovir triphosphate are present in CMV-infected than in uninfected cells. The triphosphate is a competitive inhibitor of deoxyguanosine triphosphate incorporation into DNA and preferentially inhibits viral rather than host cellular DNA polymerases. Ganciclovir is incorporated into both viral and cellular DNA. Incorporation into viral DNA causes eventual cessation of DNA chain elongation (see Figure 501B and Figure 502). A novel strategy of suicide gene therapy involves transduction of the HSV thymidine kinase gene into tumor cells by viral vectors. Subsequent exposure to ganciclovir induces apoptosis and cell-death receptor expression (Beltinger et al., 1999). Intracellular ganciclovir triphosphate concentrations are 10-fold higher than those of acyclovir triphosphate and decline much more slowly with an intracellular t1/2 of elimination exceeding 24 hours (Biron et al., 1985). These differences may account in part for ganciclovir's greater anti-CMV activity and provide the rationale for single daily doses in suppressing human CMV infections. CMV can become resistant to ganciclovir by one of two mechanisms: reduced intracellular ganciclovir phosphorylation due to mutations in the viral phosphotransferase encoded by the UL97 gene and to mutations in viral DNA polymerase (Erice, 1999). Resistant CMV clinical isolates have 4- to >20-fold increases in inhibitory concentrations. Resistance has been associated primarily with impaired phosphorylation (Stanat et al., 1991), but sometimes only with DNA polymerase mutations. Highly resistant variants have dual UL97 and polymerase mutations and are variably cross-resistant to cidofovir or foscarnet. Ganciclovir also is much less active against acyclovir-resistant, thymidine kinasedeficient HSV strains. Absorption, Distribution, and Elimination The oral bioavailability of ganciclovir averages 6% to 9% following
ingestion with food and less in the fasting state. Peak and trough plasma
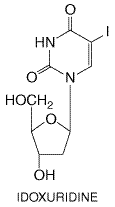
levels are about 0.5 to 1.2 The plasma half-life is about 2 to 4 hours in patients with normal renal function. Over 90% of ganciclovir is eliminated unchanged by renal excretion, which occurs by glomerular filtration and tubular secretion. Consequently, the plasma half-life increases almost linearly as creatinine clearance declines and may reach 28 to 40 hours in those with severe renal insufficiency. Untoward Effects Myelosuppression is the principal dose-limiting toxicity of ganciclovir. Neutropenia occurs in about 15% to 40% of patients and thrombocytopenia in 5% to 20% (Faulds and Heel, 1990). Neutropenia most commonly is observed during the second week of treatment and usually is reversible within 1 week of drug cessation. Persistent fatal neutropenia has occurred. Oral ganciclovir also causes neutropenia. Oral valganciclovir is associated with headache and gastrointestinal disturbance (nausea, pain, diarrhea) in addition to the toxicities associated with ganciclovir. Recombinant granulocyte colony-stimulating factor (G-CSF, filgrastim, lenograstim) may be useful in treating ganciclovir-induced neutropenia (see Chapter 54: Hematopoietic Agents: Growth Factors, Minerals, and Vitamins). CNS side effects occur in 5% to 15% of patients and range in severity from headache to behavioral changes to convulsions and coma. About one-third of patients have had to interrupt or prematurely stop therapy because of bone marrow or CNS toxicity. Infusion-related phlebitis, azotemia, anemia, rash, fever, liver function test abnormalities, nausea or vomiting, and eosinophilia also have been described. Teratogenicity, embryotoxicity, irreversible reproductive toxicity, and myelotoxicity have been observed in animals at ganciclovir dosages comparable to those used in human beings. Zidovudine (Hochster et al., 1990) and probably other cytotoxic agents increase the risk of myelosuppression, as do nephrotoxic agents that impair ganciclovir excretion. Probenecid and possibly acyclovir reduce renal clearance of ganciclovir. Zalcitabine increases oral ganciclovir exposure by an average of 22%. Oral ganciclovir increases the absorption and peak plasma concentrations of didanosine by approximately twofold and that of zidovudine by about 20% (see Chapter 51: Antiretroviral Agents: Antiretroviral Agents for discussion of zidovudine, zalcitabine, and didanosine). Therapeutic Uses Ganciclovir is effective for treatment and chronic suppression of CMV retinitis in immunocompromised patients and prevention of CMV disease in transplant patients. In CMV retinitis, initial induction treatment (5 mg/kg with food every 12 hours for 10 to 21 days) is associated with improvement or stabilization in about 85% of patients (Faulds and Heel, 1990; Drew, 1992). Reduced viral excretion is usually evident by 1 week, and funduscopic improvement by 2 weeks. Because of the high risk of relapse, AIDS patients with retinitis require suppressive therapy with high doses of ganciclovir (30 to 35 mg/kg per week). Oral ganciclovir (1000 mg three times daily) is effective for suppression of retinitis after initial intravenous treatment. Oral valganciclovir is comparable to intravenous dosing for initial control and sustained suppression of CMV retinitis. Intravitreal ganciclovir injections have been used in some patients, and an intraocular sustained-release ganciclovir implant (VITRASERT) is more effective than systemic dosing in suppressing retinitis progression (Musch et al., 1997). Ganciclovir therapy (5 mg/kg per 12 hours for 14 to 21 days) may benefit other CMV syndromes in AIDS patients or solid-organ transplant recipients (Nichols and Boeckh, 2000). Response rates of 67% or higher have been found in combination with a decrease in immunosuppressive therapy. Recurrent CMV disease occurs commonly after initial treatment. In bone-marrow transplant recipients with CMV pneumonia or gastrointestinal infection, ganciclovir alone is ineffective. However, ganciclovir combined with intravenous immunoglobulin or CMV immunoglobulin reduces the mortality of CMV pneumonia by about one-half. Ganciclovir treatment (12 mg/kg per day in 2 divided doses for 6 weeks) is associated with suppression of viuria and possibly clinical benefit in infants with congenital CMV disease (Whitley et al., 1997). Ganciclovir has been used both for prophylaxis and for suppression of CMV infections in transplant recipients. In bone-marrow transplant recipients, preemptive ganciclovir treatment (5 mg/kg per 12 hours for 7 to 14 days followed by 5 mg/kg per day to day 100 to 120 posttransplant), starting when CMV is isolated from bronchoalveolar lavage (Schmidt et al., 1991) or from other sites (Goodrich et al., 1991), is highly effective in preventing CMV pneumonia and appears to reduce mortality in these patients. Guidelines for patient monitoring (CMV blood levels, antigenemia) and use of ganciclovir prophylaxis have been published recently (Centers for Disease Control and Prevention, 2000). Initiation of ganciclovir at the time of engraftment also reduces CMV disease rates but does not improve survival, in part because of infections due to ganciclovir-related neutropenia (Goodrich et al., 1993). Preemptive therapy (5 mg/kg two times daily for 7 days) when CMV shedding occurs also appears to be effective in solid-organ transplants or during rejection episodes (Singh et al., 1994). Intravenous ganciclovir administration reduces the risk of CMV disease in solid-organ transplant recipients (Pillay, 2000). Oral ganciclovir (1000 mg three times daily for 3 months) reduces CMV disease risk in liver transplant recipients, including high-risk patients with primary infection or those receiving antilymphocyte antibodies (Gane et al., 1997). Oral ganciclovir prophylaxis is more effective than high-dose oral acyclovir in solid-organ transplant recipients (Flechner et al., 1998). In advanced HIV disease, oral ganciclovir (1000 mg three times daily) may reduce the risk of CMV disease and possibly mortality in those not receiving didanosine (Spector et al., 1996; Brosgart et al., 1998). The addition of oral high-dose ganciclovir (1500 mg three times daily) to the intraocular ganciclovir implant further delays the time to retinitis progression and reduces the risk of new CMV disease (Martin et al., 1999) and the risk of Kaposi's sarcoma. The susceptibility of strains recovered before and after therapy in transplant patients generally is unchanged, although resistance emergence occurs in a minority of patients and is associated with poorer prognosis (Kruger et al., 1999). The use of antithymocyte globulin and prolonged ganciclovir exposure are risk factors. Recovery of ganciclovir-resistant CMV isolates has been associated with progressive CMV disease in AIDS and other immunocompromised patients (Erice, 1999). Over one-quarter of retinitis patients have resistant isolates by 9 months of therapy, and resistant CMV has been recovered from cerebrospinal fluid (CSF), vitreous fluid, and visceral sites. A ganciclovir ophthalmic gel formulation appears to be effective in treating HSV keratitis (Colin et al., 1997). Oral ganciclovir reduces hepatitis B virus (HBV) DNA levels and aminotransferase levels in chronic hepatitis B (Hadziyannis et al., 1999). Systemic ganciclovir is being used in conjunction with suicide gene therapy expressing HSV thymidine kinase for treatment of brain tumors and a variety of other malignancies (Packer et al., 2000). Idoxuridine Chemistry and Antiviral Activity Idoxuridine (5-iodo-2'-deoxyuridine) is an iodinated thymidine analog that inhibits the in vitro replication of various DNA viruses, including herpesviruses and poxviruses (Prusoff, 1988). Its structure is given below:
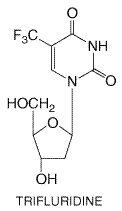
Inhibitory concentrations for HSV-1 are 2 to 10 Mechanism of Action and Resistance The antiviral mechanism of idoxuridine is not completely defined, but the phosphorylated derivatives interfere with various enzyme systems. The triphosphate inhibits viral DNA synthesis and is incorporated into both viral and cellular DNA. Such altered DNA is more susceptible to breakage and also leads to faulty transcription. Resistance to idoxuridine readily develops in vitro and occurs in viral isolates recovered from idoxuridine-treated patients with HSV keratitis. Therapeutic Uses In the Trifluridine Trifluridine (5-trifluoromethyl-2'-deoxyuridine) is a fluorinated pyrimidine nucleoside that has in vitro inhibitory activity against HSV types 1 and 2, CMV, vaccinia, and, to a lesser extent, certain adenoviruses (Carmine et al., 1982). Its structure is given below:
Concentrations of trifluridine of 0.2 to 10 Mechanism of Action and Resistance The antiviral mechanism of trifluridine involves inhibition of viral DNA synthesis. Trifluridine monophosphate irreversibly inhibits thymidylate synthetase, and trifluridine triphosphate is a competitive inhibitor of thymidine triphosphate incorporation into DNA by DNA polymerases (Carmine et al., 1982). Trifluridine is incorporated into viral and cellular DNA. Trifluridine-resistant HSV with altered thymidine kinase substrate specificity can be selected in vitro, and resistance in clinical isolates has been described. Therapeutic Uses Trifluridine currently is approved in the Vidarabine Vidarabine
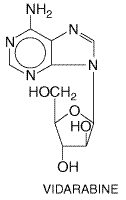
It is active against herpesviruses, poxviruses, rhabdoviruses,
hepadnaviruses, and some RNA tumor viruses (Whitley et al., 1980).
Inhibitory concentrations are 3.0 The antiviral mechanism of vidarabine is incompletely understood, but vidarabine is an inhibitor of viral DNA synthesis. Cellular enzymes phosphorylate vidarabine to the triphosphate, which inhibits viral DNA polymerase activity in a manner that is competitive with deoxyadenosine triphosphate. Vidarabine triphosphate is incorporated into both cellular and viral DNA, where it may act as a chain terminator. Vidarabine triphosphate also inhibits ribonucleoside reductase, RNA polyadenylation, and S-adenosylhomocysteine hydrolase (SAHH), an enzyme involved in transmethylation reactions. Resistant variants due to mutations in viral DNA polymerase can be selected in vitro. Intravenous vidarabine causes dose-related gastrointestinal toxicity, acute neurotoxicities, painful peripheral neuropathy, weakness, hypokalemia, rash, elevated transaminases, anemia, and leukopenia or thrombocytopenia. Vidarabine is teratogenic and oncogenic in animals. Intravenous vidarabine once was used for treating HSV encephalitis, neonatal herpes, and zoster or varicella in immunocompromised patients, but acyclovir has replaced it for these indications. Combined administration of vidarabine and acyclovir has been used occasionally in life-threatening herpesvirus infections. In HSV keratoconjunctivitis, topical vidarabine is superior to idoxuridine (Kaufman, 1988). |
Antiinfluenza Agents
|
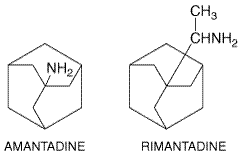
Amantadine and Rimantadine Chemistry and Antiviral Activity Amantadine
(1-adaman-tanamine hydrochloride) and its
Both agents specifically inhibit the replication of influenza A
viruses at low concentrations (Hayden and Aoki, 1999). Depending on the assay
method and strain, inhibitory concentrations of the drugs range from about
0.03 to 1.0 Mechanisms of Action and Resistance Amantadine and rimantadine share two mechanisms of antiviral action (Hayden and Aoki, 1999). They inhibit an early step in viral replication, probably viral uncoating; for some strains, they have an effect on a late step in viral assembly probably mediated through altering hemagglutinin processing. The primary locus of action is the influenza A virus M2 protein, an integral membrane protein that functions as an ion channel. By interfering with this function of the M2 protein, the drugs inhibit the acid-mediated dissociation of the ribonucleoprotein complex early in replication and potentiate acidic pH-induced conformational changes in the hemagglutinin during its intracellular transport later in replication. Resistant variants are rare (<1%) in field isolates (Zieger et al., 1999), but selected readily by virus passage in the presence of drug and have been recovered from treated persons. Resistance with over 100-fold increases in inhibitory concentrations has been associated with single nucleotide changes leading to amino acid substitutions in the transmembrane region of M2 (Hayden, 1996). Amantadine and rimantadine share cross-susceptibility and resistance. Absorption, Distribution, and Elimination Amantadine and rimantadine are well absorbed after oral administration
(see Table 504) (Aoki and Sitar, 1988; Wills et al., 1987).
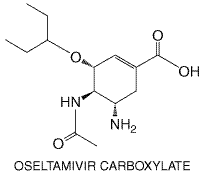
Peak plasma concentrations of amantadine average 0.5 to 0.8 Both drugs have very large volumes of distribution. Nasal secretion and salivary levels of amantadine approximate those found in the serum. Amantadine is excreted in breast milk. Rimantadine concentrations in nasal mucus average 50% higher than those in plasma. Amantadine is excreted largely unmetabolized in the urine through glomerular filtration and probably tubular secretion. The plasma t1/2 of elimination is about 12 to 18 hours in young adults. Because amantadine's elimination is highly dependent on renal function, the t1/2 of elimination increases up to twofold in the elderly and even more in those with renal impairment (Horadam et al., 1981). Dose adjustments are advisable in those with mild decrements in renal function. In contrast, rimantadine is metabolized extensively by hydroxylation, conjugation, and glucuronidation prior to renal excretion. Following oral administration, the plasma t1/2 of elimination of rimantadine averages 24 to 36 hours, and 60% to 90% is excreted in the urine as metabolites (Wills et al., 1987). Renal clearance of unchanged rimantadine is similar to creatinine clearance. Untoward Effects The most common side effects related to amantadine and rimantadine are minor dose-related gastrointestinal and CNS complaints (Hayden and Aoki, 1999). These include nervousness, lightheadedness, difficulty concentrating, insomnia, and loss of appetite or nausea. CNS side effects occur in approximately 5% to 33% of patients treated with amantadine at doses of 200 mg/day, but are significantly less frequent with rimantadine. Amantadine dose reductions are required in older adults (100 mg/day) because of decreased renal function, but 20% to 40% of infirm elderly will experience side effects even at this lower dose. At comparable doses of 100 mg per day, rimantadine is significantly better tolerated in nursing home residents than is amantadine (Keyser et al., 2000). High amantadine plasma concentrations (1.0 to 5.0 The neurotoxic effects of amantadine appear to be increased by concomitant ingestion of antihistamines and psychotropic or anticholinergic drugs, especially in the elderly. Therapeutic Uses Amantadine and rimantadine are effective for prevention and treatment of influenza A virus infections. Seasonal prophylaxis with either drug (a total of 200 mg/day in 1 or 2 divided doses in young adults) is about 70% to 90% protective against influenza A illness (Hayden and Aoki, 1999). Efficacy has been shown during pandemic influenza, in preventing nosocomial influenza, and in curtailing nosocomial outbreaks. Doses of 100 mg/day are better tolerated and appear to be protective against influenzal illness. Postexposure prophylaxis with either drug provides protection of exposed family contacts, if ill young children are not concurrently treated. Seasonal prophylaxis is an alternative in high-risk patients, if the influenza vaccine cannot be administered or may be ineffective. Prophylaxis should be started as soon as influenza is identified in a community or region and should be continued throughout the period of risk (usually 4 to 8 weeks), since any protective effects are lost several days after cessation. Alternatively, the drugs can be started in conjunction with immunization and continued for 2 weeks until protective immune responses develop. In uncomplicated influenza A illness of adults, early amantadine or rimantadine treatment (200 mg/day for 5 days) reduces the duration of fever and systemic complaints by 1 to 2 days, speeds functional recovery, and sometimes decreases the duration of virus shedding (Hayden and Aoki, 1999). In children, rimantadine treatment may be associated with less illness and lower viral titers during the first 2 days of treatment, but rimantadine-treated children have more prolonged shedding of virus. The optimal dose and duration of therapy have not been established in children for either agent. It also is uncertain whether treatment reduces risk of complications in high-risk patients or is useful in patients with established pulmonary complications. Resistant variants have been recovered from approximately 30% of treated children or adults by the fifth day of therapy (Hayden, 1996). Resistant variants also arise commonly when amantadine or rimantadine is used to treat influenza in immunocompromised patients (Englund et al., 1998). Illnesses due to apparent transmission of resistant virus, associated with failure of drug prophylaxis, have been documented in contacts of drug-treated ill persons in households and in nursing homes. Resistant variants appear to be pathogenic and can cause typical disabling influenzal illness. The discovery that amantadine also is useful in treating parkinsonism was due to serendipity. This application is discussed in Chapter 22: Treatment of Central Nervous System Degenerative Disorders. Amantadine and rimantadine have been used alone or in combination with interferon and other agents in treating chronic hepatitis C with inconsistent results to date (Younossi and Perrillo, 1999). Oseltamivir Chemistry and Antiviral Activity Oseltamivir carboxylate [(3R, 4R, 5S)-4-acetylamino-5-amino-3(1-ethylpropoxyl)-1-cyclohexene-1-carboxylic acid] is a transition-state analog of sialic acid that is a potent, selective inhibitor of influenza A and B virus neuraminidases (Kim et al., 1997). Its structure is shown below. Oseltamivir phosphate is an ethyl ester prodrug that lacks antiviral activity. Oseltamivir carboxylate has an antiviral spectrum and potency similar to that of zanamivir (see below) (Mendel et al., 1998). It inhibits amantadine- and rimantadine-resistant influenza A viruses and some zanamivir-resistant variants.
Mechanisms of Action and Resistance Influenza neuraminidase cleaves terminal sialic acid residues and destroys the receptors recognized by viral hemagglutinin, which are present on the cell surface, progeny virions, and in respiratory secretions (Gubareva et al., 2000). This enzymatic action is essential for release of virus from infected cells. Interaction of oseltamivir carboxylate with the neuraminidase causes a conformational change within the enzyme's active site and inhibition of activity. Inhibition of neuraminidase activity leads to viral aggregation at the cell surface and reduced virus spread within the respiratory tract. Influenza variants selected in vitro for resistance to oseltamivir carboxylate contain hemagglutinin and/or neuraminidase mutations (McKimm-Breschkin, 2000). Resistance has not been recognized in influenza B viruses to date. The most commonly recognized variants (mutations at positions 292 or 274 of neuraminidase) have reduced infectivity and virulence in vivo. Oral oseltamivir therapy has been associated with recovery of resistant variants in about 1% to 2% of treated adults. Absorption, Distribution, and Elimination Oral oseltamivir phosphate is rapidly absorbed (about 80%; see Table
504) and cleaved by esterases in the gastrointestinal tract or liver to the
antivirally active carboxylate. Low blood levels of oseltamivir phosphate are
detectable, but are only 3% to 5% of those of the metabolite. The
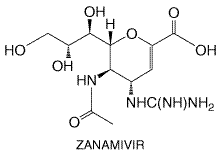
bioavailability of the carboxylate is estimated to be Untoward Effects Oral oseltamivir is associated with nausea, abdominal discomfort, and, less often, emesis, probably due to local irritation. Gastrointestinal complaints usually are mild to moderate in intensity, typically resolve despite continued dosing in 1 to 2 days, and are preventable by administration with food. The frequency of such complaints is about 10% to 15% when oseltamivir is used for treatment of influenza illness and less than 5% when used for prophylaxis. An increased frequency of headache was reported in one prophylaxis study in elderly adults. Oseltamivir phosphate and the carboxylate do interact with the cytochrome P450 system in vitro. Their protein binding is low. No clinically significant drug interactions have been recognized to date. High doses of oseltamivir cause renal tubular mineralization and delayed parturition in mice; these effects are of uncertain clinical significance. Therapeutic Uses Oral oseltamivir is effective in the treatment and prevention of influenza. Treatment of previously healthy adults (75 mg twice daily for 5 days) or children aged 1 to 12 years (2 mg/kg twice daily for 5 days) with acute influenza reduces illness duration by about 1 to 2 days, speeds functional recovery, and reduces the risk of complications leading to antibiotic use by 40% to 50% (Treanor et al., 2000; Whitley et al., 2001). Efficacy in the elderly and in high-risk patients with underlying cardiopulmonary conditions is under study. When used for prophylaxis during the influenza season, oseltamivir (75 mg once daily) is effective in reducing the likelihood of influenza illness in both unimmunized working adults and in immunized nursing-home residents (Hayden et al., 1999; Peters et al., 1999), and short-term use (7 days) protects against influenza in household contacts. Zanamivir Chemistry and Antiviral Activity Zanamivir (4-guanidino-2,4-dideoxy-2,3-dehydro-N-acetyl neuraminic acid) is a sialic acid analog that potently and specifically inhibits the neuraminidases of influenza A and B viruses (von Itzstein et al., 1993). Its structure is shown below. Depending on the strain, zanamivir competitively inhibits influenza neuraminidase activity at concentrations of approximately 0.2 to 3 ng/ml (Woods et al., 1993) but affects neuraminidases from other pathogens and mammalian sources only at 106-fold higher concentrations. Zanamivir inhibits in vitro replication of influenza A and B viruses, including amantadine- and rimantadine-resistant strains, and is active after topical administration in animal models of influenza.
Mechanisms of Action and Resistance Like oseltamivir, zanamivir inhibits viral neuraminidase and thus causes viral aggregation at the cell surface and reduced spread of virus within the respiratory tract (Gubareva et al., 2000). In vitro selection of viruses resistant to zanamivir is associated with mutations in the viral hemagglutinin and/or neuraminidase (McKimm-Breschkin, 2000). Hemagglutinin variants generally have mutations in or near the receptor binding site that make them less dependent on neuraminidase action for release from cells in vitro, although they may retain susceptibility in vivo (Woods et al., 1993). Hemagglutinin variants are cross-resistant to other neuraminidase inhibitors. Neuraminidase variants contain mutations in the enzyme active site that diminish binding of zanamivir, but the altered enzymes show reduced activity or stability. Resistant variants may have decreased infectivity in animals. Resistance emergence has not been documented with zanamivir in immunocompetent hosts to date. One resistant influenza B variant containing dual hemagglutinin and neuraminidase mutations was recovered from an immunocompromised child treated with nebulized zanamivir (Gubareva et al., 1998). Absorption, Distribution, and Elimination The oral bioavailability of zanamivir is low (<5%; see Table 504), and most clinical trials have used intranasal or dry powder inhalation delivery. The proprietary inhaler device for delivering zanamivir in a lactose carrier is breath-actuated and requires a cooperative patient. Following inhalation of the dry powder, approximately 15% is deposited in the lower respiratory tract and about 80% in the oropharynx (Cass et al., 1999). Overall bioavailability is less than 20%, and plasma levels after 10-mg inhaled doses average about 35 to 100 ng/ml in adults and children (Peng et al., 2000a). Median zanamivir concentrations in induced sputum samples are 1336 ng/ml at 6 hours and 47 ng/ml at 24 hours after a single 10-mg dose in healthy volunteers (Peng et al., 2000b). The plasma half-life of zanamivir averages 2.5 to 5 hours after oral inhalation but only 1.7 hours following intravenous dosing. Over 90% is eliminated in the urine without recognized metabolism. Untoward Effects Topically applied zanamivir generally is well tolerated in ambulatory adults and children with influenza. Wheezing and bronchospasm have been reported in some influenza-infected patients without known airway disease, and acute deteriorations in lung function, including fatal outcomes, have occurred in those with underlying asthma or chronic obstructive airway disease. No significant changes in lung function or airway reactivity were found in uninfected mild to moderate asthmatics given 2 weeks of inhaled zanamivir (Cass et al., 2000). Tolerability in more serious bronchopulmonary disorders or in intubated patients is uncertain. Zanamivir administration to patients with underlying airway disease requires close monitoring and availability of rapidly acting bronchodilators and should be stopped if problems develop. Preclinical studies of zanamivir revealed no evidence of mutagenic, teratogenic, or oncogenic effects. No clinically significant drug interactions have been recognized to date. Zanamivir does not diminish the immune response to injected influenza vaccine. Therapeutic Uses Inhaled zanamivir is effective for prevention and treatment of acute influenza. Early zanamivir treatment (10 mg twice daily for 5 days) of febrile influenza in ambulatory adults and children aged 5 years and older shortens the time to illness resolution by 1 to 3 days (Hayden et al., 1997; Hedrick et al., 2000). In previously healthy adults, zanamivir treatment also reduces by 40% the risk of lower respiratory tract complications leading to antibiotic use. Once-daily inhaled, but not intranasal, zanamivir is highly protective against community-acquired influenza illness (Monto et al., 1999), and when given for 10 days, it protects against household transmission (Hayden et al., 2000). Intravenous zanamivir is protective against experimental human influenza but has not been studied in treating natural influenza. |
Other Antiviral Agents
|
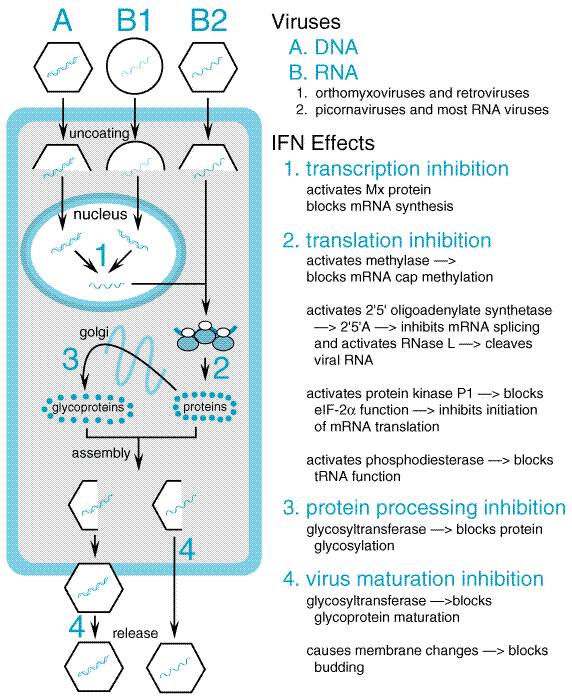
Interferons Classification and Antiviral Activity Interferons (IFNs) are potent cytokines that possess antiviral, immunomodulating, and antiproliferative actions (Baron et al., 1992; see also Chapter 53: Immunomodulators: Immunosuppressive Agents, Tolerogens, and Immunostimulants). These proteins are synthesized by cells in response to various inducers and in turn cause biochemical changes leading to an antiviral state in cells of the same species. Three major classes of human interferons with significant antiviral activity currently are recognized: alpha (>18 individual species), beta, and gamma. Clinically used recombinant alpha interferons (Table 502) are nonglycosylated proteins of approximately 19,500 daltons. Preparations of natural and recombinant interferons alpha available for clinical use are referred to as interferons alfa. Interferon alpha and interferon beta may be produced by nearly all cells in response to viral infection and a variety of other stimuli, including double-stranded RNA and certain cytokines (e.g., interleukin 1, interleukin 2, and tumor necrosis factor). Interferon gamma production is restricted to T lymphocytes and natural killer cells responding to antigenic stimuli, mitogens, and specific cytokines. Interferons alpha and beta exhibit antiviral and antiproliferative actions; stimulate the cytotoxic activity of lymphocytes, natural killer cells, and macrophages; and upregulate class I major histocompatibility antigens (MHC) and other surface markers. Interferon gamma has less antiviral activity but more potent immunoregulatory effects, particularly macrophage activation, expression of class II MHC, and mediation of local inflammatory responses. Most animal viruses are inhibited by the antiviral actions of interferons, although many DNA viruses are relatively insensitive. Considerable differences in potency exist among different viruses and assay systems. Interferon biological activity usually is measured in terms of antiviral effects in cell culture and generally is expressed as international units (IU) relative to reference standards. Mechanisms of Action Following binding to specific cellular receptors, interferons activate the JAK-STAT signal transduction pathway and lead to the nuclear translocation of a cellular protein complex that binds to genes containing an interferon-specific response element. This, in turn, leads to synthesis of over two dozen proteins that contribute to viral resistance (Stark et al., 1998; Figure 503). The antiviral effects of interferon are mediated through inhibition of viral penetration or uncoating, synthesis of messenger RNA, translation of viral proteins, and/or viral assembly and release. Inhibition of protein synthesis is the major inhibitory effect for many viruses. Interferon-induced proteins include 2'-5'-oligoadenylate [2-5(A)] synthetases and a protein kinase, either of which can inhibit protein synthesis in the presence of double-stranded RNA. The 2-5(A) synthetase produces adenylate oligomers that activate a latent cellular endoribonuclease (RNase L) to cleave both cellular and viral single-stranded RNAs. The protein kinase selectively phosphorylates and inactivates a protein involved in protein synthesis, eukaryotic initiation factor 2 (eIF-2). Interferon-induced protein kinase also may be an important effector of apoptosis. Interferon also induces a phosphodiesterase, which cleaves a portion of transfer RNA and thus prevents peptide elongation. A particular virus may be inhibited at several steps, and the principal inhibitory effect for a specific virus differs among virus families. In addition, certain viruses are able to counter interferon effects by blocking production or activity of selected interferon-inducible proteins. For example, interferon resistance in hepatitis C virus is attributable to inhibition of protein kinase and to other mechanisms (Francois et al., 2000).
Complex interactions exist between interferons and other parts of the immune system. Interferons may ameliorate viral infections by exerting direct antiviral effects and/or by modifying the immune response to infection. For example, interferon-induced expression of major histocompatibility antigens may contribute to the antiviral actions of interferon by enhancing the lytic effects of cytotoxic T lymphocytes. In addition to contributing to controlling infection, interferons may mediate some of the systemic symptoms associated with viral infections and contribute to immunologically mediated tissue damage in certain viral diseases. Absorption, Distribution, and Elimination Oral administration does not result in detectable interferon levels in serum or increases in 2-5(A) synthetase activity in peripheral blood mononuclear cells (Wills, 1990). After intramuscular or subcutaneous injection of interferon alfa, absorption exceeds 80%. Plasma levels are dose-related, peaking at 4 to 8 hours and returning to baseline by 18 to 36 hours. Levels of 2-5(A) synthetase in peripheral-blood mononuclear cells, which have been used as a marker of interferon's biologic activity, show increases beginning at 6 hours and lasting through 4 days after a single injection. An antiviral state in peripheral-blood mononuclear cells peaks at 24 hours and slowly decreases to baseline by 6 days after injection. Absorption of interferon gamma is more variable, and intramuscular or subcutaneous injections of interferon beta result in negligible plasma levels, although increases in 2-5(A) synthetase levels may occur. The volume of distribution of interferon alfa averages about 31 liters. After systemic administration, low levels of interferon are detected in respiratory secretions, CSF, eye, and brain. Because interferons induce long-lasting biological effects, their activities are not easily predictable from usual pharmacokinetic measures. After intravenous dosing, clearance of interferon from plasma occurs in a complex, multiexponential manner (Bocci, 1992). The plasma elimination half-life of interferon alfa is about 40 minutes; those of recombinant interferon beta or interferon gamma are approximately 4 hours and 0.5 hour, respectively. Elimination from the blood relates to distribution to the tissues, cellular uptake, and catabolism primarily in the kidney and liver. Negligible amounts are excreted in the urine. Attachment of interferon proteins to large inert polyethylene glycol (PEG) molecules (pegylation) decreases the clearance substantially. Plasma concentrations of interferon are prolonged, and the extended duration of therapeutic activity allows for once-weekly dosing. Pegylation also may increase the antigenicity of the protein to which it is bonded. Two pegylated interferons have received extensive clinical testing. Peginterferon alfa-2b has a straight chain, 12,000-dalton type of PEG which increases the half-life from approximately 2 to 3 hours to about 54 hours (Glue et al., 2000). Peginterferon alfa-2a consists of an ester derivative of a branched-chain 40,000-dalton PEG bonded to interferon alfa-2a and has a plasma half-life averaging 77 hours. Increasing PEG size is associated with longer half-life and less renal clearance and relative antiviral activity. About 70% of PEG interferon alfa-2b is cleared by hepatic metabolism; peginterferon alfa-2a also is cleared primarily by the liver. Untoward Effects Injection of interferon doses of 1 to 2 million units (MU) or greater usually is associated with an acute influenzalike syndrome beginning several hours after injection. Symptoms include fever, chills, headache, myalgia, arthralgia, nausea, vomiting, and diarrhea (Dusheiko, 1997). Fever usually resolves within 12 hours. Tolerance gradually develops in most patients. Febrile responses can be moderated by pretreatment with various antipyretics. Up to one-half of patients receiving intralesional therapy for genital warts experience the influenzal illness initially, as well as discomfort at the injection site, and leukopenia. The principal dose-limiting toxicities of systemic interferon are myelosuppression with granulocytopenia and thrombocytopenia; neurotoxicity manifested by somnolence, confusion, behavioral disturbance, and rarely seizures; debilitating neurasthenia with fatigue and weight loss; autoimmune disorders including thyroiditis; and, uncommonly, cardiovascular effects with hypotension and tachycardia. Elevations in hepatic enzymes and triglycerides, alopecia, proteinuria and azotemia, interstitial nephritis, autoantibody formation, and hepatotoxicity may occur. Alopecia and personality change are common in interferon-treated children (Sokal et al., 1998). The development of serum neutralizing antibodies to exogenous interferons may be associated infrequently with Newer Agents under Clinical Development
loss of clinical responsiveness (Antonelli et al., 1991). Interferon may impair fertility, and safety during pregnancy is not established. Interferon reduces the metabolism of various drugs by the hepatic cytochrome P450 system and significantly increases levels of drugs such as theophylline. Interferons can increase the bone-marrow toxicity of myelotoxic drugs such as zidovudine. Pegylated interferons are tolerated as well as standard interferons with discontinuation rates ranging from 6% to 11%, although the frequencies of fever, nausea, and injection-site inflammation appear to be somewhat higher in some studies. The safety of PEG accumulation and long-term circulation has not been established. Therapeutic Uses Recombinant, natural, and pegylated alpha interferons (Table 502)
currently are approved in the Hepatitis B Virus In patients with chronic hepatitis B, parenteral administration of various interferons is associated with loss of hepatitis B virus (HBV) DNA, loss of HBV e antigen (HBeAg), and development of anti-HBe antibody, and biochemical and histological improvement in about 25% to 50% of the patients (Haria and Benfield, 1995; Main and Thomas, 1997). Lasting responses require moderately high interferon doses and prolonged administration (typically 5 MU/day or 10 MU in adults and 6 MU/m2 in children three times per week for 4 to 6 months) (Sokal et al., 1998). Plasma HBV DNA and polymerase activity decline promptly in most patients, but complete disappearance is sustained in only about one-third. Low pretherapy serum HBV DNA levels and high aminotransferase levels are predictors of response. Sustained responses are infrequent in those with vertically acquired infection, anti-HBe positivity, or concurrent immunosuppression due to HIV. Responses with seroconversion to anti-HBe usually are associated with transaminase elevations and often a hepatitis-like illness during the second or third month of therapy, likely related to immune clearance of infected hepatocytes. High-dose interferon can cause myelosuppression and clinical deterioration in those with decompensated liver disease. Remissions in chronic hepatitis B induced by interferon are sustained in over 80% of patients treated and frequently are followed by loss of HBV surface antigen (HbsAg), histological improvement or stabilization, and reduced risk of liver-related complications and mortality (Lau et al., 1997). Interferon may benefit HBV-associated nephrotic syndrome and glomerulonephritis in some patients. Antiviral effects and improvements occur in about one-half of chronic hepatitis D virus (HDV) infections, but relapse is common unless HbsAg disappears (Farci et al., 1994). Interferon does not appear to be beneficial in acute HBV or hepatitis D virus (HDV) infections. Hepatitis C Virus In chronic HCV infection, subcutaneous interferon alfa-2b monotherapy (3 MU three times a week) is associated with an approximate 50% to 70% rate of aminotransferase normalization and loss of plasma viral RNA. However, relapse rates are high, and sustained virologic remission is observed in only about 10% to 25% of patients treated for 6 months (Main and Thomas, 1997). Prolonged treatment (12 to 18 months) and possibly higher doses increase the likelihood of sustained responses. Sustained responses are associated with long-term histologic improvement and possibly reduced risk of hepatocellular carcinoma (Yoshida et al., 1999). Viral genotype and pretreatment RNA level influence response to treatment, but early viral clearance is the best predictor of sustained response (Civeira and Prieto, 1999). Those who are HCV RNAnegative at 3 months of initiating therapy generally should continue treatment for 12 months or longer (Gish, 1999). Pegylated interferons are superior to conventional thrice-weekly
interferon monotherapy in inducing sustained remissions in treatment-nave
patients. A once- weekly dosing regimen of peginterferon alfa-2a (180 Nonresponders generally do not benefit from interferon monotherapy retreatment but may respond to combined interferon and ribavirin therapy (see'Ribavirin,' below). Patients relapsing after monotherapy may respond to interferon retreatment or more often to combined interferon-ribavirin therapy. Interferon treatment may benefit HCVassociated cryoglobulinemia and glomerulonephritis. Interferon administration during acute hepatitis C infection appears to reduce the risk of chronicity. Papillomavirus In refractory condylomata acuminata (genital warts), intralesional injection of various natural and recombinant interferons is associated with complete clearance of injected warts in 36% to 62% of patients (Frazer and McMillan, 1997). Relapse occurs in 20% to 30% of patients with interferon-induced remission. Verruca vulgaris may respond to intralesional interferon alfa. Intramuscular or subcutaneous administration is associated with some regression in wart size but greater toxicity, and no higher complete response rate when used as an adjunctive modality. Systemic interferon may provide adjunctive benefit in recurrent juvenile laryngeal papillomatosis and in treating laryngeal disease in older patients. Other Viruses Interferons have been shown to have virologic and/or clinical effects in various herpesvirus infections including genital HSV infections, localized herpes zoster of cancer patients or of older adults, and CMV infections of renal transplant patients. However, interferon generally is associated with more side effects and inferior clinical benefits compared to conventional antiviral therapies. Topically applied interferon and trifluridine combinations appear active in drug-resistant mucocutaneous HSV infections (Birch et al., 1992). In HIV-infected persons, interferons have been associated with antiretroviral effects. In advanced infection, however, the combination of zidovudine and interferon is associated with only transient benefit and excessive hematological toxicity. Interferon alfa (3 MU three times weekly) is effective for treatment of HIV-related thrombocytopenia resistant to zidovudine therapy (Marroni et al., 1994). Except for adenovirus, interferon has broad-spectrum antiviral activity against respiratory viruses in vitro. However, prophylactic intranasal interferon alfa is protective only against rhinovirus colds, and chronic use is limited by the occurrence of nasal side effects. Intranasal interferon is therapeutically ineffective in established rhinovirus colds. Lamivudine Lamivudine, the () enantiomer of 2',3'-dideoxy-3'thiacytidine, is a nucleoside analog that inhibits HIV reverse transcriptase and HBV DNA polymerase. Its use as an antiretroviral agent is discussed in depth in Chapter 51: Antiretroviral Agents: Antiretroviral Agents. It inhibits HBV replication in vitro by 50% at concentrations of 4 to 7 ng/ml with negligible cellular cytotoxicity. Cellular enzymes convert lamivudine to the triphosphate, which competitively inhibits HBV DNA polymerase and causes chain termination. The intracellular t1/2 of the triphosphate averages 17 to 19 hours in HBV-infected cells, so that infrequent dosing is possible. HBV resistance to lamivudine is associated with from 40- to 104-fold reduced in vitro susceptibility. Resistant HBV variants recovered from treated patients have mutations in the viral DNA polymerase, particularly involving position 550/2 in the YMDD motif and often position 526/8. Some variants appear to replicate less efficiently than wild-type virus in vitro, and some are lamivudine-dependent (Yeh et al., 2000). Following oral administration, lamivudine is rapidly absorbed with a bioavailability of about 80% in adults (Johnson et al., 1999). Peak plasma levels are observed at 0.5 to 1.5 hours after dosing and average approximately 1000 ng/ml after 100-mg doses. Lamivudine is distributed widely in a volume comparable to total body water. The plasma t1/2 averages about 9 hours, and approximately 70% of the dose is excreted unchanged in the urine. About 5% is metabolized to an inactive trans-sulfoxide metabolite. In HBV-infected children, doses of 3 mg/kg per day provide plasma exposure and trough plasma levels comparable to those in adults receiving 100 mg daily (Sokal et al., 2000). Dose reductions are indicated for moderate renal insufficiency (creatinine clearance <50 ml/minute). Trimethoprim decreases the renal clearance of lamivudine. Long-term lamivudine treatment (100 mg daily for Resistance emergence with return of detectable HBV DNA occurs in 14% to 32% of immunocompetent patients treated with lamivudine (100 mg daily) by 1 year and increases over time to over 50% by three years of therapy. The clinical significance of genotypic resistance is under study. Virologic breakthroughs are often subclinical but may be associated with clinical and biochemical deterioration. Despite higher doses of lamivudine (300 mg daily), dually infected HIV patients also have high frequencies of resistance emergence. Resistance development in HBV-infected liver-transplant recipients occurs frequently and may be associated with histological worsening. Ribavirin Chemistry and Antiviral Activity Ribavirin
Ribavirin inhibits the replication of a wide range of RNA and DNA
viruses, including orthomyxo-, paramyxo-, arena-, bunya-, flavi-, herpes-,
adeno-, pox-, and retroviruses (Gilbert and Knight, 1986; Huggins, 1989). In
vitro inhibitory concentrations range from 3 to 10 Mechanisms of Action and Resistance The antiviral mechanism of ribavirin is not fully defined but relates to alteration of cellular nucleotide pools and inhibition of viral messenger RNA synthesis (Gilbert and Knight, 1986). Intracellular phosphorylation to the mono-, di-, and triphosphate derivatives is mediated by host cell enzymes. In both uninfected and RSV-infected cells, the predominant derivative (>80%) is the triphosphate, which has an intracellular t1/2 of elimination of less than 2 hours. Ribavirin monophosphate competitively inhibits cellular inosine-5'-phosphate dehydrogenase and interferes with the synthesis of guanosine triphosphate (GTP) and thus nucleic acid synthesis in general. Ribavirin triphosphate also competitively inhibits the GTP-dependent 5'-capping of viral messenger RNA, and specifically influenza virus transcriptase activity. Ribavirin appears to have multiple sites of action, and some of these (e.g., inhibition of GTP synthesis) may potentiate others (e.g., inhibition of GTP-dependent enzymes). Emergence of viral resistance to ribavirin has not been documented in clinical isolates, although it has been possible to select cells that do not phosphorylate it to active forms. Absorption, Distribution, and Elimination Ribavirin is actively taken up by gastrointestinal nucleoside
transporters located in the proximal small bowel, and oral bioavailability
averages approximately 50% (Glue, 1999). Extensive accumulation occurs in
plasma, and steady state is reached by about 4 weeks. Food increases plasma
levels substantially, so ingestion with food may be prudent (Glue, 1999).
Following single or multiple oral doses of 600 mg and 1200 mg, peak plasma
concentrations average 0.8 The apparent volume of distribution is large ( Untoward Effects Aerosolized ribavirin may cause mild conjunctival irritation, rash, transient wheezing, and occasional reversible deterioration in pulmonary function. When used in conjunction with mechanical ventilation, equipment modifications and frequent monitoring are required to prevent plugging of ventilator valves and tubing with ribavirin. Techniques to reduce environmental exposure of health care workers are important (Shults et al., 1996). Systemic ribavirin causes dose-related reversible anemia due to extravascular hemolysis and suppression of bone marrow (Huggins, 1989). Associated increases occur in reticulocyte counts and in serum bilirubin, iron, and uric acid concentrations. High ribavirin triphosphate levels may cause oxidative damage to membranes, leading to erythrophagocytosis by the reticuloendothelial system (De Franceschi et al., 2000). Bolus intravenous infusion may cause rigors. About 20% of chronic hepatitis C patients receiving combination interferon-ribavirin therapy discontinue treatment early because of side effects. In addition to interferon toxicity, oral ribavirin increases the risk of fatigue, cough, rash, pruritus, nausea, insomnia, dyspnea, depression, and, particularly, anemia. About 8% of patients require ribavirin dose reduction because of anemia. Preclinical studies indicate that ribavirin is teratogenic, embryotoxic, oncogenic, and possibly gonadotoxic. To prevent possible teratogenic effects, up to 6 months is required for washout following cessation of long-term treatment (Glue, 1999). Pregnant women should not directly care for patients receiving ribavirin aerosol. Ribavirin is in FDA pregnancy category X. Ribavirin inhibits the phosphorylation and antiviral activity of pyrimidine nucleoside HIV reverse-transcriptase inhibitors such as zidovudine and stavudine but increases the activity of purine nucleoside reverse-transcriptase inhibitors (e.g., didanosine) in vitro. Therapeutic Uses Ribavirin aerosol is approved in the Oral ribavirin in combination with injected interferon-alfa (REBETRON) is effective for treatment of chronic hepatitis C. Ribavirin monotherapy for 6 to 12 months reversibly decreases aminotransferase elevations to normal in about 30% of patients but does not affect HCV RNA levels. Combination therapy with interferon-alfa (3 million units subcutaneously three times weekly) and oral ribavirin (500 mg, or 600 mg twice daily if weight is greater than 75 kg for 24 to 48 weeks) increases the likelihood of sustained biochemical and virologic responses to about 40% depending on genotype (Battaglia and Hagmeyer, 2000). The combination is superior to interferon alone in both treatment-nave patients (McHutchison and Poynard, 1999) and in those not responding to or relapsing after interferon monotherapy (Barbaro et al., 1999). A longer duration of therapy (48 weeks) appears to benefit those with genotype 1 infections, high plasma HCV-RNA levels, or advanced fibrosis. Combined therapy has been used in the management of recurrent HCV infection after liver transplantation (Lavezzo and Rizzetto, 1999). Intravenous and/or aerosol ribavirin has been used occasionally in treating severe influenza virus infection and in the treatment of immunosuppressed patients with adenovirus, vaccinia, parainfluenza, or measles virus infections. Aerosolized ribavirin is associated with reduced duration of fever but no other clinical or antiviral effects in influenza infections in hospitalized children (Rodriguez et al., 1994). Intravenous ribavirin decreases mortality in Lassa fever and has been used in treating other arenavirus-related hemorrhagic fevers. In hemorrhagic fever with renal syndrome due to Hantaan virus infection (Huggins et al., 1991), intravenous ribavirin is beneficial, and it is under study in hantavirus-associated pulmonary syndrome. Oral ribavirin has been used for treatment of Crimean-Congo hemorrhagic fever. Intravenous ribavirin is investigational in the United States. Imiquimod Imiquimod
(1-(2-methylpropyl)-1H-imidazo[4,5-c]quino-lin-4 amine) is a novel
immunomodulatory agent that is effective for topical treatment of condylomata
acuminata (Miller et al., 1999). In vitro, it lacks direct
antiviral or antiproliferative effects but rather induces interferon-alpha,
tumor necrosis factor-alpha (TNF- |
Prospectus
|
More satisfactory antiviral therapies likely will come in part from the identification of agents with improved pharmacokinetic properties, greater potency, and/or improved toxicity profiles compared to existing ones. New drug-delivery techniques that improve pharmacokinetic properties or target particular tissues also will be of benefit. Prodrugs that can be used to enhance oral absorption and/or avoid degradation of the parent compound are receiving particular attention in drug development. As in other areas of antimicrobial chemotherapy, the combined use of antiviral agents has been studied as a means of increasing antiviral activity, reducing drug dosage and the associated risk of toxicity, and preventing or modifying the development of drug resistance. Because viral isolates may be mixtures of sensitive and resistant viruses or viruses with different resistance mutations, treatment with combinations of drugs may provide broader activity than treatment with single agents. Drug combinations may constrain the mutability of the virus, enhance susceptibility to a second agent, or diminish viral replicative capacity. Future therapeutic breakthroughs probably will depend on the identification of novel molecular targets in viruses. A particularly interesting area of investigation is gene inhibition therapy (e.g., antisense oligonucleotides, ribozymes). This approach not only may inhibit active replication but potentially may eradicate latent viral infection. The first antisense oligonucleotide has been approved for a human viral infection (fomivirsen for CMV retinitis), but important problems regarding potency, selectivity, and pharmacology remain to be solved. Interesting approaches to gene therapy include expression of mutated proteins that act as transdominant inhibitors and intracellular expression of antibody fragments against critical viral proteins. Other approaches that may prove to be useful involve agents to moderate host immunopathological responses, agents to boost host immune responses, or virus-specific immunotherapies (e.g., monoclonal antibodies, therapeutic vaccines) to supplement host responses. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1536
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved