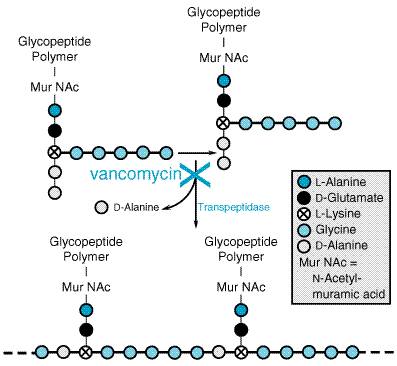
| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antimicrobial Agents: Protein Synthesis Inhibitors and Miscellaneous Antibacterial Agents
Overview
|
The antimicrobial agents discussed in this
chapter are: (1) bacteriostatic, protein-synthesis inhibitors that act
principally by binding to ribosomes; (2) non- |
Tetracyclines
|
History Tetracycline antibiotics were discovered by systematic screening of soil specimens collected from many parts of the world for antibiotic-producing microorganisms. The first of these compounds, chlortetracycline, was introduced in 1948. Tetracyclines were found to be highly effective against rickettsiae, a number of gram-positive and gram-negative bacteria, and Chlamydia, and hence became known as 'broad-spectrum' antibiotics. With establishment of their in vitro antimicrobial activity, effectiveness in experimental infections, and pharmacological properties, the tetracyclines rapidly became widely used in therapy. Although there are specific and useful differences among the
tetracyclines currently available in the Source and Chemistry Chlortetracycline and oxytetracycline are elaborated by Streptomyces aureofaciens and Streptomyces rimosus, respectively. Tetracycline is produced semisynthetically from chlortetracycline. Demeclocycline is the product of a mutant strain of Strep. aureofaciens, and methacycline, doxycycline, and minocycline are all semisynthetic derivatives. The tetracyclines are close congeners of polycyclic naphthacenecarboxamide. Their structural formulas are shown in Table 471. Effects on Microorganisms The tetracyclines are active against a wide range of aerobic and
anaerobic gram-positive and gram-negative bacteria. They also are effective
against some microorganisms that are resistant to cell-wall-active
antimicrobial agents, such as Rickettsia, Coxiella burnetii, Mycoplasma
pneumoniae, Chlamydia spp., Legionella spp., Ureaplasma,
some atypical mycobacteria, and Plasmodium spp. They are not active
against fungi. Demeclocycline, tetracycline, oxytetracycline, minocycline,
and doxycycline are available in the The more lipophilic drugs, minocycline and doxycycline, usually are
the most active by weight, followed by tetracycline. Resistance of a
bacterial strain to any one member of the class usually results in
cross-resistance to other tetracyclines. Most bacterial strains that are
inhibited by Bacteria In general, tetracyclines are more active against gram-positive than gram-negative microorganisms. Problems of resistance and the availability of superior antimicrobial agents limit the use of tetracyclines for treatment of infections caused by many gram-positive bacteria. Most strains of enterococci are resistant to tetracycline; group B streptococci are 50% susceptible, and only 65% or less of Staphylococcus aureus remain susceptible (Standiford, 2000). Both tetracycline and doxycycline are quite active against most strains of S. pneumoniae, although penicillin-resistant strains also are often resistant to tetracyclines (Doern et al., 1998). Although the tetracyclines initially were useful for treatment of infections with aerobic gram-negative organisms, many Enterobacteriaceae are now relatively resistant. However, more than 90% of strains of H. influenzae still may be sensitive to doxycycline (Doern et al., 1997). Although all strains of Pseudomonas aeruginosa are resistant, 90% of strains of Pseudomonas pseudomallei (the cause of melioidosis) are sensitive. Most strains of Brucella also are susceptible. Tetracyclines are particularly useful for infections caused by Haemophilus ducreyi (chancroid), Brucella, and Vibrio cholerae. These drugs also inhibit the growth of Legionella pneumophila, Campylobacter jejuni, Helicobacter pylori, Yersinia pestis, Yersinia enterocolitica, Francisella tularensis, and Pasteurella multocida. Strains of N. gonorrhoeae and Neisseria meningitidis, once uniformly susceptible to tetracycline, generally are resistant (Harnett et al., 1997). The tetracyclines are active against many anaerobic and facultative
microorganisms, and their activity against Actinomyces is particularly
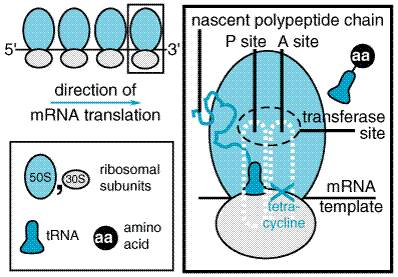
relevant. The MIC breakpoint for susceptible anaerobic bacteria is 8 Rickettsiae Like chloramphenicol, all of the tetracyclines are highly effective against the rickettsiae responsible for Rocky Mountain spotted fever, murine typhus, epidemic typhus, scrub typhus, rickettsialpox, and Q fever (C. burnetii). Miscellaneous Microorganisms The tetracyclines are active against many spirochetes, including Borrelia recurrentis, Borrelia burgdorferi (Lyme disease), Treponema pallidum (syphilis), and Treponema pertenue. The activity of tetracyclines against Chlamydia and Mycoplasma has become particularly important. Strains of Mycobacterium marinum also are susceptible. Effects on Intestinal Flora Many of the tetracyclines are incompletely absorbed from the gastrointestinal tract, such that high concentrations are reached in the bowel, and therefore the enteric flora is markedly altered. Many aerobic and anaerobic coliform microorganisms and gram-positive spore-forming bacteria are sensitive and may be suppressed markedly during long-term tetracycline regimens before resistant strains reappear. The stools become softer and odorless and acquire a yellow-green color. However, as the fecal coliform count declines, overgrowth of tetracycline-resistant microorganisms occurs, particularly of yeasts (Candida spp.), enterococci, Proteus, and Pseudomonas. Tetracycline occasionally produces pseudomembranous colitis caused by toxin from Clostridium difficile. Mechanism of Action Tetracyclines inhibit bacterial protein synthesis by binding to the 30 S bacterial ribosome and preventing access of aminoacyl tRNA to the acceptor (A) site on the mRNA-ribosome complex (see Figure 471). They enter gram-negative bacteria by passive diffusion through the hydrophilic channels formed by the porin proteins of the outer cell membrane, and active transport by an energy-dependent system that pumps all tetracyclines across cytoplasmic membrane. Although permeation of these drugs into gram-positive bacteria is less well understood, it also is energy requiring.
At high concentrations, these compounds impair protein synthesis in mammalian cells. However, because mammalian cells lack the active transport system found in bacteria, and the ribosomal target is less sensitive, tetracyclines are selectively active against bacteria. Resistance to the Tetracyclines Microorganisms that have become resistant to one tetracycline frequently are resistant to the others. Resistance to the tetracyclines in Escherichia coli and probably in other bacterial species is primarily plasmid-mediated and is an inducible trait. The three main resistance mechanisms are: (1) decreased accumulation of tetracycline as a result of either decreased antibiotic influx or acquisition of an energy-dependent efflux pathway; (2) decreased access of tetracycline to the ribosome because of the presence of ribosome protection proteins; and (3) enzymatic inactivation of tetracyclines (Speer et al., 1992). Absorption, Distribution, and Excretion Absorption Absorption of most tetracyclines from the gastrointestinal tract is incomplete. The percentage of an oral dose that is absorbed (when the stomach is empty) is lowest for chlortetracycline (30%); intermediate for oxytetracycline, demeclocycline, and tetracycline (60% to 80%); and high for doxycycline (95%) and minocycline (100%) (Barza and Scheife, 1977). The percentage of unabsorbed drug rises as the dose increases. Most absorption takes place from the stomach and upper small intestine and is greater in the fasting state. Absorption of tetracyclines is impaired by the concurrent ingestion of dairy products; aluminum hydroxide gels; calcium, magnesium, and iron or zinc salts; and bismuth subsalicylate. The mechanism responsible for the decreased absorption appears to be chelation of divalent and trivalent cations. The wide range of plasma concentrations present in different individuals following the oral administration of the various tetracyclines is related to the variability of their absorption. These drugs can be divided into three groups based on the dosage and frequency of oral administration required to produce effective plasma concentrations. Oxytetracycline and tetracycline are incompletely absorbed. After a
single oral dose, the peak plasma concentration is attained in 2 to 4 hours.
These drugs have half-lives in the range of 6 to 12 hours and are frequently
administered two to four times daily. The administration of 250 mg every 6
hours produces peak plasma concentrations of 2 to 2.5 Demeclocycline, which also is incompletely absorbed, usually is administered in lower daily dosages than are the above-mentioned congeners, because its half-life of about 16 hours permits effective plasma concentrations lasting for 24 to 48 hours. Doxycycline and minocycline should be administered in even lower daily
dosages by the oral route, since their half-lives are long (16 to 18 hours)
and they are better absorbed (90% to 100%) than tetracycline, oxytetracycline,
or demeclocycline. After an oral dose of 200 mg of doxycycline, a maximum
plasma concentration of 3 Distribution Tetracyclines distribute widely throughout the body and into tissues and secretions, including the urine and prostate. They accumulate in the reticuloendothelial cells of the liver, spleen, and bone marrow, and in bone, dentine, and the enamel of unerupted teeth (see below). Inflammation of the meninges is not a prerequisite for the passage of tetracyclines into the cerebrospinal fluid (CSF). Penetration of these drugs into most other fluids and tissues is excellent. Concentrations in synovial fluid and the mucosa of the maxillary sinus approach that in plasma. Tetracyclines cross the placenta and enter the fetal circulation and amniotic fluid. Concentrations of tetracycline in umbilical-cord plasma reach 60%, and in amniotic fluid 20%, of those in the circulation of the mother. Relatively high concentrations of these drugs also are found in breast milk. Excretion The primary route of elimination for most tetracyclines is the kidney (doxycycline being an important exception), although they are also concentrated in the liver and excreted by way of the bile into the intestines, where they are partially reabsorbed via enterohepatic recirculation. Elimination via the intestinal tract occurs even when the drugs are given parenterally, as a result of excretion into the bile. Minocycline is an exception and is significantly metabolized by the liver. Since renal clearance of these drugs is by glomerular filtration, their excretion is significantly affected by the renal function status of the patient (see below). From 20% to 60% of an intravenous 0.5-g dose of tetracycline is excreted in the urine during the first 24 hours; from 20% to 55% of an oral dose is excreted by this route. Approximately 10% to 35% of a dose of oxytetracycline is excreted in active form in the urine, in which it is detectable within 30 minutes and reaches a peak concentration about 5 hours after it is administered. The rate of renal clearance of demeclocycline is less than half that of tetracycline. About 50% of methacycline is excreted unchanged in the urine. Decreased hepatic function or obstruction of the common bile duct reduces the biliary excretion of these agents, resulting in longer half-lives and higher plasma concentrations. Because of their enterohepatic circulation, the tetracyclines may be present in the body for a long time after cessation of therapy. Minocycline is recoverable from both urine and feces in significantly lower amounts than are the other tetracyclines, and it appears to be metabolized to a considerable extent. Renal clearance of minocycline is low. The drug persists in the body after its administration is stopped; this may be due to retention in fatty tissues. The half-life of minocycline is not prolonged in patients with hepatic failure. With conventional doses, doxycycline is not eliminated via the same pathways as are other tetracyclines, and it does not accumulate significantly in patients with renal failure. It is thus one of the safest of the tetracyclines for the treatment of extrarenal infections in such individuals. The drug is excreted in the feces, largely as an inactive conjugate or perhaps as a chelate; for this reason it has less impact on the intestinal microflora (Nord and Heimdahl, 1988). The half-life of doxycycline may be shortened from approximately 16 to 7 hours in patients who are receiving long-term treatment with barbiturates or phenytoin. Routes of Administration and Dosage The tetracyclines are available in a wide variety of forms for oral,
parenteral, and topical administration. As indicated earlier, only tetracycline
(ACHROMYCIN, others), oxytetracycline (TERRAMYCIN, others), demeclocycline (DECLOMYCIN), minocycline (MINOCIN, others), doxycycline (VIBRAMYCIN, others), and chlortetracycline (AUREOMYCIN) are available in the Oral Administration The appropriate oral dose of the tetracyclines varies with the nature and the severity of the infection being treated. For tetracycline, it ranges from 1 to 2 g per day in adults. Children over 8 years of age should receive 25 to 50 mg/kg daily in two to four divided doses. The recommended dose of demeclocycline is somewhat lower, being 150 mg every 6 hours or 300 mg every 12 hours for adults. The daily dose for children over 8 years of age is 6 to 12 mg/kg in two to four divided portions. Demeclocycline, however, is rarely used as an antimicrobial agent because of its higher risks of photosensitivity reactions and diabetes insipidus syndrome (see below). The dose of doxycycline for adults is 100 mg every 12 hours during the first 24 hours, followed by 100 mg once a day, or twice daily when severe infection is present. Children over 8 years of age should receive doxycycline, 4 to 5 mg/kg per day, divided into two equal doses given every 12 hours the first day, after which half this amount (2 to 2.5 mg/kg) should be given as a single daily dose. In serious disease, the 2 to 2.5 mg/kg dose is given every 12 hours. The dose of minocycline for adults is 200 mg initially, followed by 100 mg every 12 hours; for children it is 4 mg/kg initially, followed by 2 mg/kg every 12 hours. Gastrointestinal distress, nausea, and vomiting can be minimized by administration of the tetracyclines with food (but not dairy products). Dairy products; antacids containing calcium, aluminum, zinc, magnesium, or silicate; vitamins with iron; sulcralfate (which contains aluminum); and bismuth subsalicylate will chelate and therefore interfere with the absorption of tetracyclines and should not be ingested at the same time. Cholestyramine and colestipol also bind orally administered tetracyclines and interfere with their absorption. Parenteral Administration Doxycycline is the preferred parenteral tetracycline in the The usual intravenous dose of doxycycline is 200 mg in one or two
infusions on the first day and 100 to 200 mg on subsequent days. The dose for
children who weigh less than 45 kg is 4.4 mg/kg on the first day, after which
it is reduced correspondingly. The total daily dose of intravenous tetracycline
(where available) for most acute infections is 500 mg to 1 g, usually
administered in equally divided doses at 6-hour or 12-hour intervals. Up to 2
g per day may be given in severe infections. This dose should not be exceeded
and may cause difficulty in some patients (see'Toxic Effects,'
below). Parenteral preparations of tetracycline no longer are available in
the Local Application Except for local use in the eye, topical use of the tetracyclines is not recommended. Ophthalmic preparations include chlortetracycline hydrochloride, tetracycline hydrochloride, and oxytetracycline hydrochloride; they are available as ophthalmic ointments or suspensions. Their use in ophthalmic therapy is discussed in Chapter 66: Ocular Pharmacology. Therapeutic Uses The tetracyclines have been used extensively both for the treatment of infectious diseases and as an additive to animal feeds to facilitate growth. Both uses have resulted in dramatically increased bacterial resistance to these drugs, and their use has declined. Tetracyclines are especially useful in diseases caused by rickettsiae, mycoplasmas, and chlamydiae. The status of the tetracyclines for the therapy of various infections is given in Table 431. Rickettsial Infections The tetracyclines and chloramphenicol are effective and may be life-saving in rickettsial infections, including Rocky Mountain spotted fever, recrudescent epidemic typhus (Brill's disease), murine typhus, scrub typhus, rickettsialpox, and Q fever. Clinical improvement often is evident within 24 hours after initiation of therapy. Mycoplasma Infections Mycoplasma pneumoniae is sensitive to the tetracyclines. Treatment of pneumonia with either tetracycline or erythromycin results in a shorter duration of fever, cough, malaise, fatigue, pulmonary rales, and radiological changes in the lungs. Mycoplasma may persist in the sputum following cessation of therapy, despite rapid resolution of the active infection. Chlamydia Lymphogranuloma Venereum Doxycycline (100 mg twice daily for 21 days) is first-line therapy for treatment of this infection (Prevention, 1998). Decided reduction in the size of buboes occurs within 4 days, and inclusion and elementary bodies entirely disappear from the lymph nodes within 1 week. Lymphogranulomatous proctitis is improved promptly. Rectal pain, discharge, and bleeding are decreased markedly. When relapses occur, treatment is resumed with full doses and is continued for longer periods. Pneumonia, bronchitis, or sinusitis caused by Chlamydia pneumoniae responds to tetracycline therapy. The tetracyclines also are of value in cases of psittacosis. Drug therapy for 10 to 14 days usually is adequate. Trachoma Doxycycline (100 mg twice daily for 14 days) or tetracycline (250 mg four times daily for 14 days) is effective for this infection. However, this disease is important in early childhood, and tetracyclines therefore often are contraindicated (see'Untoward Effects,' below). Azithromycin (see'Macrolides (Erythromycin, Clarithromycin, and Azithromycin)'), which is effective as a single dose, is preferred. Nonspecific Urethritis Nonspecific urethritis is often due to Chlamydia trachomatis. One hundred mg of doxycycline every 12 hours for 7 days is effective, although azithromycin, which can be given as a single 1-g dose, is preferred because of improved compliance. Sexually Transmitted Diseases Tetracyclines have been effective for uncomplicated gonococcal
infections. Doxycycline (100 mg twice daily for 7 days) is still recommended
for treatment of gonorrhea, although cefixime, ceftriaxone (see Chapter
45: Antimicrobial Agents: Penicillins, Cephalosporins, and Other C. trachomatis often is a coexistent pathogen in acute pelvic inflammatory disease,
including endometritis, salpingitis, parametritis, and/or peritonitis (Walker
et al., 1993). Doxycycline, 100 mg intravenously twice daily, is
recommended for at least 48 hours after substantial clinical improvement,
followed by oral therapy at the same dosage to complete a 14-day course.
Doxycycline usually is combined with cefoxitin or cefotetan (seeChapter
45: Antimicrobial Agents: Penicillins, Cephalosporins, and Other Acute epididymitis is caused by infection with C. trachomatis or N. gonorrhoeae in men less than 35 years of age. Effective regimens include a single injection of ceftriaxone (250 mg) plus doxycycline, 100 mg orally twice daily for 10 days. Sexual partners of patients with any of the above conditions should also be treated. Nonpregnant, penicillin-allergic patients who have primary, secondary, or latent syphilis can be treated with a tetracycline regimen such as doxycycline 100 mg orally twice daily for 2 weeks (Centers for Disease Control and Prevention, 1998). Tetracyclines should not be used for treatment of neurosyphilis. Bacillary Infections Brucellosis Tetracyclines are effective for acute and chronic infections caused by Brucella melitensis, Brucella suis, and Brucella abortus. Combination therapy with doxycycline, 200 mg per day, plus rifampin (seeChapter 48: Antimicrobial Agents: Drugs Used in the Chemotherapy of Tuberculosis, Mycobacterium avium Complex Disease, and Leprosy), 600 to 900 mg daily for 6 weeks, is recommended by the World Health Organization for the treatment of acute brucellosis (World Health Organization, 1986). Relapses usually respond to a second course of therapy. The combination of doxycycline given with streptomycin (1 g daily, intramuscularly) also is effective and may be more efficacious than doxy-cycline-rifampin in patients with spondylitis ( Ariza et al., 1992). Tularemia Although streptomycin (seeChapter 48: Antimicrobial Agents: Drugs Used in the Chemotherapy of Tuberculosis, Mycobacterium avium Complex Disease, and Leprosy) is preferable, treatment with the tetracyclines also produces prompt results in tularemia. Both the ulceroglandular and typhoidal types of the disease respond well. Fever, toxemia, and clinical signs and symptoms all are improved. Cholera Doxycycline (300 mg as a single dose) is effective in reducing stool volume and eradicating Vibrio cholerae from the stool within 48 hours. Antimicrobial agents, however, are not substitutes for fluid and electrolyte replacement in this disease. In addition, some strains of V. cholerae are resistant to tetracyclines (Khan et al., 1996). Other Bacillary Infections Therapy with the tetracyclines is often ineffective in infections caused by Shigella, Salmonella, or other Enterobacteriaceae because of a high prevalence of drug-resistant strains in many areas. Doxycycline has been used successfully to reduce the incidence of travelers' diarrhea, but a high prevalence of resistance in enteric bacteria limits the usefulness of the drug for this indication. Coccal Infections Because of the emergence of resistance, the tetracyclines are no longer indicated for infections caused by staphylococci, streptococci, or meningococci. Approximately 85% of strains of S. pneumoniae are susceptible to tetracyclines. Doxycycline remains an effective agent for empirical therapy of community-acquired pneumonia (Ailani et al., 1999; Bartlett et al., 1998). Urinary Tract Infections Tetracyclines are no longer recommended for routine treatment of urinary tract infections, because many enteric organisms, including E. coli, that cause these infections are resistant. Other Infections Actinomycosis, although most responsive to penicillin G, may be successfully treated with a tetracycline. Minocycline has been suggested as an alternative for the treatment of nocardiosis, but a sulfonamide should be used concurrently. Yaws and relapsing fever respond favorably to the tetracyclines. Tetracyclines have been shown to be useful in the acute treatment and for prophylaxis of leptospirosis (Leptospira spp.). Borrelia spp., including B. recurrentis (relapsing fever) and B. burgdorferi (Lyme disease), respond to therapy with a tetracycline. The tetracyclines have been used to treat atypical mycobacterial pathogens when susceptible, including M. marinum. Acne Tetracyclines have been used for the treatment of acne. These drugs may act by inhibiting propionibacteria, which reside in sebaceous follicles and metabolize lipids into irritating free fatty acids. Tetracycline seems to be associated with few side effects when given in relatively low doses of 250 mg orally twice a day. Untoward Effects Toxic Effects Gastrointestinal The tetracyclines all produce gastrointestinal irritation to varying degrees in some but not all individuals; such effects are more common after oral administration of the drugs. Epigastric burning and distress, abdominal discomfort, nausea, and vomiting may occur. Gastric distress can be reduced by administering the drug with food, but tetracyclines should not be taken with dairy products. Esophagitis and esophageal ulcers have been reported (Winckler, 1981; Amendola and Spera, 1985), as has an association with pancreatitis (Elmore and Rogge, 1981). Diarrhea also may result from the irritative effects of the tetracyclines given orally. Pseudomembranous colitis caused by overgrowth of Clostridium difficile is a potentially life-threatening complication (see below). Photosensitivity Demeclocycline, doxycycline, and, to a lesser extent, other derivatives may produce mild to severe photosensitivity reactions in the skin of treated individuals exposed to sunlight. Onycholysis and pigmentation of the nails may develop with or without accompanying photosensitivity. Hepatic Toxicity Oxytetracycline and tetracycline appear to be the least hepatotoxic of these agents. Most hepatic toxicity develops in patients receiving 2 g or more of drug per day parenterally; however, this effect also may occur when large quantities are administered orally. Pregnant women appear to be particularly susceptible to severe tetracycline-induced hepatic damage. Jaundice appears first, and azotemia, acidosis, and irreversible shock may follow. Renal Toxicity Tetracyclines may aggravate uremia in patients with renal disease by inhibiting protein synthesis and provoking a catabolic effect. Doxycycline has fewer renal side effects than do other tetracyclines. Nephrogenic diabetes insipidus has been observed in some patients receiving demeclocycline, and this phenomenon has been exploited for the treatment of chronic inappropriate secretion of antidiuretic hormone (see Chapter 30:Vasopressin and Other Agents Affecting the Renal Conservation of Water). A clinical syndrome characterized by nausea, vomiting, polyuria, polydipsia, proteinuria, acidosis, glycosuria, and gross aminoaciduriaa form of the Fanconi syndromehas been observed in patients ingesting outdated and degraded tetracycline. It results from a toxic effect on proximal renal tubules. Effects on Teeth Children receiving long- or short-term therapy with a tetracycline may develop brown discoloration of the teeth. The larger the dose of drug relative to body weight, the more intense the discoloration of enamel. This discoloration is permanent. The duration of therapy appears to be less important than the total quantity of antibiotic administered. The risk of this untoward effect is highest when the tetracycline is given to neonates and babies prior to the first dentition. However, pigmentation of the permanent dentition may develop if the drug is given between the ages of 2 months and 5 years, when these teeth are being calcified. The deposition of the drug in the teeth and bones probably is due to its chelating property and the formation of a tetracyclinecalcium orthophosphate complex. Treatment of pregnant patients with tetracyclines may produce discoloration of the teeth in their children. The period of greatest danger to the teeth is from midpregnancy to about 4 to 6 months of the postnatal period for the deciduous anterior teeth, and from a few months to 5 years of age for the permanent anterior teeth, the periods when the crowns of the teeth are being formed. However, children up to 8 years old may be susceptible to this complication of tetracycline therapy. Miscellaneous Effects Tetracyclines are deposited in the skeleton during gestation and throughout childhood. A 40% depression of bone growth, as determined by measurement of fibulas, has been demonstrated in premature infants treated with these agents (Cohlan et al., 1963). This depression is readily reversible if the period of exposure to the drug is short. Thrombophlebitis frequently follows intravenous administration, especially when a single vein is used for repeated infusion. This irritative effect of tetracyclines has been used therapeutically in patients with malignant pleural effusions, where drug is instilled into the pleural space. Long-term therapy with tetracyclines may produce changes in the peripheral blood. Leukocytosis, atypical lymphocytes, toxic granulation of granulocytes, and thrombocytopenic purpura have been observed. The tetracyclines may cause increased intracranial pressure and tense bulging of the fontanels (pseudotumor cerebri) in young infants, even when given in the usual therapeutic doses. Except for the elevated pressure, the spinal fluid is normal. Discontinuation of therapy results in prompt return of the pressure to normal. This complication may occur rarely in older individuals (Walters and Gubbay, 1981). Patients receiving minocycline may experience vestibular toxicity, manifested by dizziness, ataxia, nausea, and vomiting. The symptoms occur soon after the initial dose and generally disappear within 24 to 48 hours after drug administration is stopped. The frequency of this side effect is directly related to the dose, and the effect has been noted more often in women than in men (Fanning et al., 1977). Hypersensitivity Reactions Various skin reactions, including morbilliform rashes, urticaria, fixed drug eruptions, and generalized exfoliative dermatitis, may follow the use of any of the tetracyclines, but they are rare. Among the more severe allergic responses are angioedema and anaphylaxis; anaphylactoid reactions can occur even after the oral use of these agents. Other effects that have been attributed to hypersensitivity are burning of the eyes, cheilosis, atrophic or hypertrophic glossitis, pruritus ani or vulvae, and vaginitis; these effects often persist for weeks or months after cessation of tetracycline therapy. The exact cause of these reactions is unknown. Fever of varying degrees and eosinophilia may occur when these agents are administered. Asthma also has been observed. Cross-sensitization among the various tetracyclines is common. Biological Effects Other Than Allergic or Toxic Like all antimicrobial agents, the tetracyclines administered orally or parenterally may lead to the development of superinfections caused by strains of bacteria or yeasts resistant to these agents. Vaginal, oral, and even systemic infections with yeasts and fungi are observed. The incidence of these infections appears to be much higher with the tetracyclines than with the penicillins. Pseudomembranous colitis, due to an overgrowth of toxin-producing C. difficile, is characterized by severe diarrhea, fever, and stools containing shreds of mucous membrane and a large number of neutrophils. The toxin is cytotoxic to mucosal cells and causes shallow ulcerations that can be seen by sigmoidoscopy. Discontinuation of the drug, combined with the oral administration of metronidazole, usually is curative. To decrease the incidence of toxic effects, the following precautions should be observed in the use of the tetracyclines. They should not be given to pregnant patients; they should not be employed for treatment of common infections in children under the age of 8 years; and unused supplies of these antibiotics should be discarded. |
Chloramphenicol
|
History and Source Chloramphenicol is an antibiotic produced by Streptomyces venezuelae, an
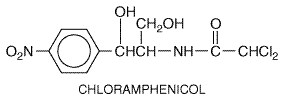
organism first isolated in 1947 from a soil sample collected in Chemistry Chloramphenicol has the following structural formula:
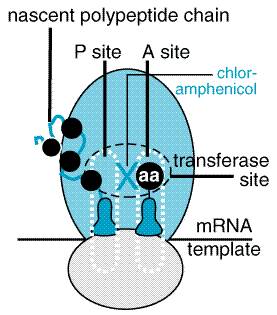
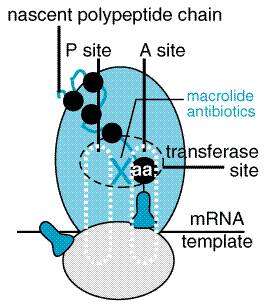
The antibiotic is unique among natural compounds in that it contains a nitrobenzene moiety and is a derivative of dichloroacetic acid. The biologically active form is levorotatory. Mechanism of Action Chloramphenicol inhibits protein synthesis in bacteria and, to a lesser extent, in eukaryotic cells. The drug readily penetrates bacterial cells, probably by facilitated diffusion. Chloramphenicol acts primarily by binding reversibly to the 50 S ribosomal subunit (near the site of action of the macrolide antibiotics and clindamycin, which it inhibits competitively). Although binding of tRNA at the codon recognition site on the 30 S ribosomal subunit is thus undisturbed, the drug appears to prevent the binding of the amino acidcontaining end of the aminoacyl tRNA to the acceptor site on the 50 S ribosomal subunit. The interaction between peptidyltransferase and its amino acid substrate cannot occur, and peptide bond formation is inhibited (seeFigure 472).
Chloramphenicol also can inhibit mitochondrial protein synthesis in mammalian cells, perhaps because mitochondrial ribosomes resemble bacterial ribosomes (both are 70 S) more than they do the 80 S cytoplasmic ribosomes of mammalian cells. The peptidyltransferase of mitochondrial ribosomes, but not cytoplasmic ribosomes, is susceptible to the inhibitory action of chloramphenicol. Mammalian erythropoietic cells seem to be particularly sensitive to the drug. Antimicrobial Actions Chloramphenicol possesses a wide spectrum of antimicrobial activity.
Strains are considered sensitive if they are inhibited by concentrations of 8
The Enterobacteriaceae have a variable sensitivity to chloramphenicol.
Most strains of E. coli (75% or more) and Klebsiella pneumoniae
are susceptible. Approximately 50% of strains of Proteus mirabilis and
indole-positive Proteus spp. are susceptible (Standiford, 2000). P.
aeruginosa is resistant to even very high concentrations of
chloramphenicol. Strains of V. cholerae have remained largely
susceptible to chloramphenicol. Strains of Shigella and Salmonella
resistant to multiple drugs, including chloramphenicol, are on the rise (Prats
et al., 2000; Replogle et al., 2000). Of special concern is the
increasing prevalence of multiple-drug-resistant strains of Salmonella
serotype typhi, particularly for strains acquired outside the Resistance to Chloramphenicol Resistance to chloramphenicol usually is caused by a plasmid-encoded
acetyltransferase that inactivates the drug. At least three types of enzyme
have been characterized (Gaffney and Foster, 1978). Acetylated derivatives of
chloramphenicol fail to bind to bacterial ribosomes. Plasmid-mediated
resistance to chloramphenicol in Salmonella serotype typhi first
emerged as a significant problem during the epidemic of 1972 through 1973 in Absorption, Distribution, Fate, and Excretion Chloramphenicol (CHLOROMYCETIN) has been available for oral administration
in two forms: the active drug itself and the inactive prodrug,
chloramphenicol palmitate (which was used to prepare an oral suspension). The
palmitate form no longer is available in the The preparation of chloramphenicol for parenteral use is the water-soluble, inactive prodrug sodium succinate preparation (chloramphenicol succinate). Similar concentrations of chloramphenicol succinate in plasma are achieved after intravenous and intramuscular administration (Shann et al., 1985). It is unclear where the hydrolysis of chloramphenicol succinate occurs in vivo, but esterases of the liver, kidneys, and lungs all may be involved. Chloramphenicol succinate itself is rapidly cleared from plasma by the kidneys. This renal clearance of the prodrug may affect the overall bioavailability of chloramphenicol, because up to 20% to 30% of the dose may be excreted prior to hydrolysis. Poor renal function in the neonate and other states of renal insufficiency result in increased plasma concentrations of chloramphenicol succinate and of chloramphenicol (Slaughter et al., 1980; Mulhall et al., 1983). Decreased esterase activity has been observed in the plasma of neonates and infants. This results in a prolonged period to reach peak concentrations of active chloramphenicol (up to 4 hours) and a longer period over which renal clearance of chloramphenicol succinate can occur. Chloramphenicol is well distributed in body fluids and readily reaches therapeutic concentrations in CSF, where values are approximately 60% of those in plasma (range, 45% to 99%) in the presence or absence of meningitis (Friedman et al., 1979). The drug actually may accumulate in brain tissue (Kramer et al., 1969). Chloramphenicol is present in bile, is secreted into milk, and readily traverses the placental barrier. It also penetrates into the aqueous humor after subconjunctival injection. The major route of elimination of chloramphenicol is hepatic metabolism to the inactive glucuronide. This metabolite, as well as chloramphenicol itself, is excreted in the urine by filtration and secretion. Over a 24-hour period, 75% to 90% of an orally administered dose is so excreted; about 5% to 10% is in the biologically active form. Patients with hepatic cirrhosis or otherwise impaired hepatic function have decreased metabolic clearance, and dosage should be adjusted in these individuals. The half-life of chloramphenicol has been correlated with plasma bilirubin concentrations (Koup et al., 1979). About 50% of chloramphenicol is bound to plasma proteins; such binding is reduced in cirrhotic patients and in neonates. The half-life of the active drug (4 hours) is not altered significantly by renal insufficiency, and dosage adjustment is not required. The extent to which hemodialysis removes chloramphenicol from plasma does not appear to warrant adjustment of dosage. However, if the dose of chloramphenicol has been reduced because of cirrhosis, clearance in the patient receiving hemodialysis may be significant. This effect can be avoided by administering the maintenance dosing at the end of hemodialysis. The variability in the metabolism and pharmacokinetic parameters of chloramphenicol in neonates, infants, and children necessitates monitoring of drug concentrations in plasma, especially when an agent that enhances its metabolism (e.g., phenobarbital, phenytoin, or rifampin) is administered concomitantly (McCracken et al., 1987). Therapeutic Uses Therapy with chloramphenicol must be limited to infections for which the benefits of the drug outweigh the risks of the potential toxicities. When other antimicrobial drugs are available that are equally effective and potentially less toxic than chloramphenicol, they should be used (Standiford, 2000). Typhoid Fever Although chloramphenicol is an important drug for the treatment of typhoid fever and other types of systemic salmonella infections, other, safer drugs are available. Also, infections and epidemics in developing countries have been due to strains of Salmonella serotype typhi highly resistant to chloramphenicol (Miller et al., 1995; Ackers, 2000). Third-generation cephalosporins and quinolones are drugs of choice for the treatment of this disease. The adult dose of chloramphenicol for typhoid fever is 1 g every 6 hours for 4 weeks. Although both intravenous and oral routes have been used, the response is more rapid with oral administration. Relapses usually respond satisfactorily to retreatment; microorganisms isolated during recurrences are usually still sensitive to the antibiotic in vitro. Bacterial Meningitis Treatment with chloramphenicol produces excellent results in H.
influenzae meningitis, equal to or better than those achieved with ampicillin
(Jones and Hanson, 1977; Koskiniemi et al., 1978). Although
chloramphenicol is bacteriostatic against most microorganisms, it is
bactericidal for many meningeal pathogens, such as H. influenzae (Rahal
and Simberkoff, 1979). The total daily dose for children should be 50 to 75
mg per kilogram of body weight, divided into four equal doses given intravenously
every 6 hours for 2 weeks. However, third-generation cephalosporins are less
toxic and have replaced chloramphenicol as initial therapy for meningitis
when H. influenzae is suspected. Chloramphenicol remains an
alternative drug for the treatment of meningitis caused by N. meningitidis
and S. pneumoniae in patients who have severe allergy to Anaerobic Infections Chloramphenicol is quite effective against most anaerobic bacteria, including Bacteroides spp. It is effective for treatment of serious intraabdominal infections or brain abscesses, both of which are commonly caused by anaerobes. However, numerous equally effective and less toxic alternatives are available, and chloramphenicol is rarely indicated. Rickettsial Diseases The tetracyclines usually are the preferred agents for the treatment of rickettsial diseases. However, in patients sensitized to these drugs, in those with reduced renal function, in pregnant women, in children less than 8 years of age, and in certain patients who require parenteral therapy because of severe illness, chloramphenicol is the drug of choice. Either tetracycline or chloramphenicol produces a favorable clinical response early in the course of Rocky Mountain spotted fever (Saah, 1995). Epidemic, murine, scrub, and recrudescent typhus as well as Q fever respond well to chloramphenicol. The same dose schedule is applicable in all the rickettsial diseases. For adults, 50 mg/kg per day is recommended. Oral therapy is preferable whenever possible. The daily dose of chloramphenicol for children with these diseases is 75 mg per kilogram of body weight, divided into equal portions and given every 6 to 8 hours; if chloramphenicol palmitate is used, the daily maintenance dose may be as high as 100 mg/kg, given at the same intervals. Therapy should be continued until the general condition has improved and fever has been absent for 24 to 48 hours. The duration of illness and the incidence of relapses and complications are greatly reduced. Brucellosis Chloramphenicol is not as effective as the tetracyclines in the treatment of brucellosis. When a tetracycline is contraindicated, 750 mg to 1 g of chloramphenicol orally every 6 hours may produce a beneficial effect in both the acute and chronic forms of the disease. Relapses usually respond to retreatment. Untoward Effects Chloramphenicol inhibits the synthesis of proteins of the inner mitochondrial membrane that are synthesized in mitochondria, probably by inhibiting the ribosomal peptidyltransferase. These include subunits of cytochrome c oxidase, ubiquinone-cytochrome c reductase, and the proton-translocating ATPase. Much of the toxicity observed with this drug can be attributed to these effects. Hypersensitivity Reactions Although relatively uncommon, macular or vesicular skin rashes occur as a result of hypersensitivity to chloramphenicol. Fever may appear simultaneously or be the sole manifestation. Angioedema is a rare complication. Jarisch-Herxheimer reactions have been observed shortly after institution of chloramphenicol therapy for syphilis, brucellosis, and typhoid fever. Hematological Toxicity The most important adverse effect of chloramphenicol is on the bone marrow. Chloramphenicol affects the hematopoietic system in two ways: by a dose-related toxic effect that presents as anemia, leukopenia, or thrombocytopenia, and by an idiosyncratic response manifested by aplastic anemia, leading in many cases to fatal pancytopenia. The latter response is not dose-related. It seems to occur more commonly in individuals who undergo prolonged therapy and especially in those who are exposed to the drug on more than one occasion. A genetic predisposition is suggested by the occurrence of pancytopenia in identical twins. Although the incidence of the reaction is low1 in approximately 30,000 or more courses of therapythe fatality rate is high when bone-marrow aplasia is complete, and there is a higher risk of acute leukemia in those who recover (Shu et al., 1987). Aplastic anemia accounts for approximately 70% of cases of blood dyscrasias due to chloramphenicol. Hypoplastic anemia, agranulocytosis, thrombocytopenia, and bone-marrow inhibition made up the remainder. Absence of reported instances of aplastic anemia following parenteral administration of chloramphenicol suggested that absorption of a toxic breakdown product from the gastrointestinal tract might be responsible (Holt, 1967). Subsequently, a few cases of aplastic anemia have been described in patients who received parenteral chloramphenicol. However, some of these patients also had received other drugs known to affect the bone marrow (phenylbutazone and glutethimide). The issue thus remains unsettled. The structural feature of chloramphenicol that is responsible for aplastic anemia is hypothesized to be the nitro group, which might be metabolized by intestinal bacteria to a toxic intermediate (Jimenez et al., 1987). However, the exact biochemical mechanism has not yet been elucidated. The risk of aplastic anemia does not contraindicate the use of chloramphenicol in situations in which it is necessary. The drug should never be used, however, in undefined situations or in diseases readily, safely, and effectively treatable with other antimicrobial agents. A second, and dose-related, toxic hematologic effect of chloramphenicol
is a common and predictable (but reversible) erythroid suppression of the
bone marrow, which is probably due to an inhibitory action of the drug on
mitochondrial protein synthesis, which in turn impairs iron incorporation
into heme (Ward, 1966). Leukopenia and thrombocytopenia also may occur. The
incidence and severity of this syndrome are related to dose. It occurs
regularly when plasma concentrations are 25 The administration of chloramphenicol in the presence of hepatic disease frequently results in depression of erythropoiesis. About one-third of patients with severe renal insufficiency exhibit the same reaction. Toxic and Irritative Effects Nausea, vomiting, unpleasant taste, diarrhea, and perineal irritation may follow the oral administration of chloramphenicol. Among the rare toxic effects produced by this antibiotic are blurring of vision and digital paresthesias. Optic neuritis occurs in 3% to 5% of children with mucoviscidosis who are given chloramphenicol; there is symmetrical loss of ganglion cells from the retina and atrophy of the fibers in the optic nerve (Godel et al., 1980). Fatal chloramphenicol toxicity may develop in neonates, especially premature babies, when they are exposed to excessive doses of the drug. The illness, the gray baby syndrome, usually begins 2 to 9 days (average, 4 days) after treatment is started. The manifestations in the first 24 hours are vomiting, refusal to suck, irregular and rapid respiration, abdominal distention, periods of cyanosis, and passage of loose, green stools. All the children are severely ill by the end of the first day and, in the next 24 hours, become flaccid, turn an ashen-gray color, and become hypothermic. A similar 'gray syndrome' condition also has been reported in adults who were accidentally given excessive quantities of the drug. Death occurs in about 40% of patients within 2 days of initial symptoms. Those who recover usually exhibit no sequelae. Two mechanisms are apparently responsible for chloramphenicol toxicity
in neonates (Craft et al., 1974): (1) failure of the drug to be
conjugated with glucuronic acid, owing to inadequate activity of glucuronyl
transferase in the liver, which is characteristic of the first 3 to 4 weeks
of life; and (2) inadequate renal excretion of unconjugated drug in the
newborn. At the time of onset of the clinical syndrome, the chloramphenicol
concentrations in plasma usually exceed 100 Chloramphenicol is removed from the blood only to a very small extent by either peritoneal dialysis or traditional hemodialysis. However, both exchange transfusion and charcoal hemoperfusion have been used to treat overdose with chloramphenicol in infants (Freundlich et al., 1983). Other organ systems that have a high rate of oxygen consumption also may be affected by the action of chloramphenicol on mitochondrial enzyme systems; encephalopathic changes have been observed (Levine et al., 1970), and cardiomyopathy also has been reported (Biancaniello et al., 1981). Drug Interactions Chloramphenicol inhibits hepatic microsomal cytochrome P450 enzymes (Halpert, 1982), and thus may prolong the half-lives of drugs that are metabolized by this system. Such drugs include warfarin, dicumarol, phenytoin, chlorpropamide, antiretroviral protease inhibitors, rifabutin, and tolbutamide. Severe toxicity and death have occurred because of failure to recognize such effects. Conversely, other drugs may alter the elimination of chloramphenicol. Chronic administration of phenobarbital or acute administration of rifampin shortens the half-life of the antibiotic, presumably because of enzyme induction, and may result in subtherapeutic concentrations of the drug. |
Macrolides (Erythromycin, Clarithromycin, and Azithromycin)
|
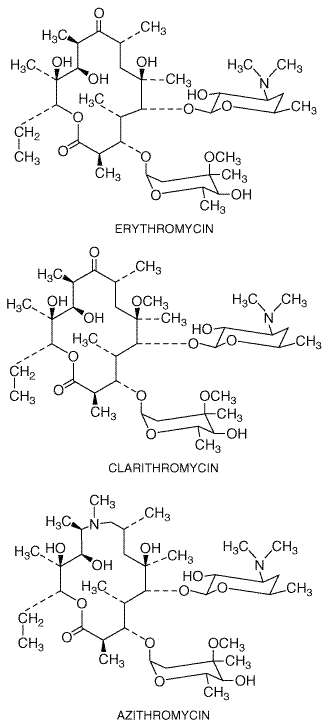
History and Source Erythromycin was discovered in 1952 by McGuire and coworkers in the metabolic products of a strain of Streptomyces erythreus, originally obtained from a soil sample collected in the Philippine archipelago. Clarithromycin and azithromycin are semisynthetic derivatives of erythromycin (Alvarez-Elcoro and Enzler, 1999). Chemistry Macrolide antibiotics, are so named because they contain a many-membered lactone ring (14-membered ring for erythromycin and clarithromycin and 15-membered ring for azithromycin) to which are attached one or more deoxy sugars. Clarithromycin differs from erythromycin only by methylation of the hydroxyl group at the 6 position, and azithromycin differs by the addition of a methyl-substituted nitrogen atom into the lactone ring. These structural modifications improve acid stability and tissue penetration and broaden the spectrum of activity. The structural formulas of the macrolides are as follows:
Antibacterial Activity Erythromycin is usually bacteriostatic, but can be bactericidal in
high concentrations against very susceptible organisms. The antibiotic is
most effective in vitro against aerobic gram-positive cocci and
bacilli (Steigbigel, 2000). Susceptible strains of S. pyogenes and S.
pneumoniae have MIC ranges from 0.015 to 1.0 Erythromycin is not active against most aerobic enteric gram-negative
bacilli. However, it has modest activity in vitro against other
gram-negative organisms, including H. influenzae (MIC, 1 to 32 Clarithromycin is more slightly potent against erythromycin-sensitive strains of streptococci and staphylococci, and has modest activity against H. influenzae and N. gonorrhoeae. Clarithromycin has good activity against M. catarrhalis, Chlamydia spp., L. pneumophila, B. burgdorferi, and Mycoplasma pneumoniae. Azithromycin generally is less active than erythromycin against gram-positive organisms (Streptococcus spp. and enterococci) and is slightly more active than either erythromycin or clarithromycin against H. influenzae and Campylobacter spp. (Peters et al., 1992). Azithromycin is very active against M. catarrhalis, P. multocida, Chlamydia spp., M. pneumoniae, L. pneumophila, B. burgdorferi, Fusobacterium spp., and N. gonorrhoeae. In general, organisms are considered susceptible to these newer agents
at a minimal inhibitory concentration (MIC breakpoint) of Both azithromycin and clarithromycin have enhanced activity against M. avium-intracellulare, as well as against some protozoa (e.g., Toxoplasma gondii, Cryptosporidium, and Plasmodium spp.). Clarithromycin has good activity against Mycobacterium leprae (Chan et al., 1994). Mechanism of Action Macrolide antibiotics are bacteriostatic agents that inhibit protein synthesis by binding reversibly to 50 S ribosomal subunits of sensitive microorganisms (Figure 473) (see Brisson-Nol et al., 1988). Erythromycin has been shown to interfere with the binding of chloramphenicol, which also acts at this site (seeFigure 472). Certain resistant microorganisms with mutational changes in components of this ribosomal subunit fail to bind the drug. It is believed that erythromycin does not inhibit peptide bond formation directly but rather inhibits the translocation step wherein a newly synthesized peptidyl tRNA molecule moves from the acceptor site on the ribosome to the peptidyl (or donor) site.
Gram-positive bacteria accumulate about 100 times more erythromycin than do gram-negative microorganisms. Cells are considerably more permeable to the nonionized form of the drug, and this fact probably explains the increased antimicrobial activity that is observed at alkaline pH (Sabath et al., 1968; Vogel et al., 1971). Acquired resistance to macrolides usually results from one of three mechanisms: (1) efflux of drug by an active pump mechanism (encoded by mrsA, mefA, or mefE in staphylococci, Group A streptococci, or S. pneumoniae, respectively); (2) inducible or constitutive production of a methylase enzyme that modifies the ribosomal target, leading to decreased drug binding, so-called ribosomal protection mediated by expression of ermA, ermB, and ermC; and (3) hydrolysis of macrolides by esterases produced by Enterobacteriaceae (Barthlmy et al., 1984). The MLSB phenotype is conferred by erm genes, indicating resistance to macrolides, lincosamides, and type B streptogramins, all of which have the same ribosomal binding site, methylase modification of which results in resistance. Chromosomal mutations that alter a 50 S ribosomal protein is a fourth mechanism of resistance found in Bacillus subtilis, Campylobacter spp., and gram-positive cocci. Absorption, Distribution, and Excretion Absorption Erythromycin base is incompletely but adequately absorbed from the
upper part of the small intestine. It is inactivated by gastric acids, and
the drug is thus administered as enteric-coated tablets or as capsules
containing enteric-coated pellets that dissolve in the duodenum. Food increases
GI acidity and may delay absorption. Peak concentrations in plasma are only
0.3 to 0.5 High concentrations of erythromycin can be achieved by intravenous
administration. Values are approximately 10 Clarithromycin is absorbed rapidly from the gastrointestinal tract
after oral administration, but its bioavailability is reduced to 50% to 55%
because of rapid first-pass metabolism. Peak concentrations occur
approximately 2 hours after drug administration. The standard formulation of
clarithromycin may be given with or without food. The extended-release form
of clarithromycin, which is given as a once-daily 1-g dose, should be
administered with food, which improves bioavailability. Steady-state peak
concentrations in plasma are 2 to 3 Azithromycin administered orally is absorbed rapidly and distributes
widely throughout the body, except to cerebrospinal fluid. Concomitant
administration of aluminum and magnesium hydroxide antacids will decrease the
peak serum drug concentrations although not the overall bioavailability;
however, it should not be administered with food. The peak plasma drug
concentration after a 500 mg loading dose is approximately 0.4 Distribution Erythromycin diffuses readily into intracellular fluids, and antibacterial activity can be achieved at essentially all sites except the brain and CSF. Erythromycin penetrates into prostatic fluid, achieving concentrations approximately 40% of those in plasma. Concentrations in middle ear exudate reach only 50% of serum concentrations, and thus may be too low for the treatment of otitis media caused by H. influenzae. Protein binding is approximately 70% to 80% for erythromycin base and even higher, 96%, for the estolate. Erythromycin traverses the placental barrier, and concentrations of the drug in fetal plasma are about 5% to 20% of those in the maternal circulation. Concentrations in breast milk also are significant (50% of those in serum). After absorption, clarithromycin undergoes rapid first-pass metabolism to its active metabolite, 14-hydroxyclarithromycin. Both of these agents distribute widely throughout the body and achieve high intracellular concentrations. Tissue concentrations generally exceed serum concentrations. Concentrations in middle ear fluid are 50% higher than simultaneous serum concentrations for both clarithromycin and the active metabolite. Protein binding of clarithromycin has been shown to range from 40% to 70% and is concentration-dependent. Azithromycin's unique pharmacokinetic properties include extensive tissue distribution and high drug concentrations within cells (including phagocytes), resulting in much greater tissue or secretion drug concentrations compared to simultaneous serum concentrations. Tissue fibroblasts act as the natural reservoir for drug in vivo, and transfer of drug to phagoctyes is easily accomplished (McDonald and Pruul, 1991). Protein binding is low (51% at very low plasma concentrations) and appears to be concentration-dependent, decreasing with increasing concentrations. Elimination Only 2% to 5% of orally administered erythromycin is excreted in active
form in the urine; this value is from 12% to 15% after intravenous infusion.
The antibiotic is concentrated in the liver and is excreted as the active
form in the bile, which may contain as much as 250 Clarithromycin is eliminated by renal and nonrenal mechanisms. It is metabolized in the liver to several metabolites, the active 14-hydroxy metabolite being the most significant. The rate of metabolism appears to be saturable and probably accounts for the nonlinear pharmacokinetics with higher dosages (Chu et al., 1992). Primary metabolic pathways are oxidative N-demethylation and stereospecific hydroxylation at the 14 position. Formation of the R- and S-epimers occurs in vivo, with the R-epimer present to a greater degree and with greater biological activity. The elimination half-lives of clarithromycin and 14-hydroxyclarithromycin are approximately 3 to 7 hours and 5 to 9 hours, respectively. Longer half-lives are observed after larger doses. The amount of clarithromycin excreted unchanged in the urine ranges from 20% to 40%, depending on the dose administered and the formulation (tablet versus oral suspension). An additional 10% to 15% of a dose is excreted in the urine as 14-hydroxyclarithromycin. Although the pharmacokinetics of clarithromycin are altered in patients with either hepatic or renal dysfunction, dosage adjustment is not necessary unless a patient has severe renal dysfunction (creatine clearance of less than 30 ml per minute). The exact biodisposition of azithromycin still is being elucidated. The drug undergoes some hepatic metabolism to inactive metabolites, but biliary excretion is the major route of elimination. Only 12% of drug is excreted unchanged in the urine. The elimination half-life, 40 to 68 hours, is prolonged because of extensive tissue sequestration and binding. Therapeutic Uses The usual oral dose of erythromycin (erythromycin base;E-MYCIN, others) for adults ranges from 1 to 2 g per day, in equally divided and spaced amounts, usually given every 6 hours, depending on the nature and severity of the infection. Daily doses of erythromycin as large as 8 g orally, given for 3 months, have been well tolerated. Food should be avoided, if possible, immediately before or after oral administration of erythromycin base or the stearate; this precaution need not be taken when erythromycin estolate (ILOSONE) or erythromycin ethylsuccinate ( E.E.S. , others) is administered. The oral dose of erythromycin for children is 30 to 50 mg/kg per day, divided into four portions; this dose may be doubled for severe infections. Intramuscular administration of erythromycin is not recommended because of pain upon injection. Intravenous administration is reserved for the therapy of severe infections, such as legionellosis. The usual dose is 0.5 to 1 g every 6 hours; 1 g of erythromycin gluceptate has been given intravenously every 6 hours for as long as 4 weeks with no difficulty except for thrombophlebitis at the site of injection. Erythromycin gluceptate (ILOTYCIN GLUCEPTATE) and erythromycinlactobionate (ERYTHROCIN LACTOBIONATE I.V.) are available for intravenous injection. Clarithromycin BIAXIN FILMTABS BIAXIN XL FILMTABS, and BIAXIN granules for suspension) usually is given as a twice-daily regimen: 250 mg twice daily for children older than 12 years and adults with mild to moderate infection. Larger doses are indicated (500 mg twice daily) for more severe infection (e.g., pneumonia) or when infection is caused by more resistant organisms (e.g., H. influenzae). Children less than 12 years old have received 7.5 mg/kg twice daily in clinical studies. The 500-mg extended-release formulation is given as two tablets once daily. Azithromycin ZITHROMAX tablet, oral suspension, and powder for intravenous injection) should be given 1 hour before or 2 hours after meals when administered orally. For outpatient therapy of community-acquired pneumonia, pharyngitis, or skin and skin-structure infections, a loading dose of 500 mg is given on the first day, then 250 mg per day is given for days 2 through 5. Treatment or prophylaxis of M. avium-intracellulare infection in AIDS patients requires higher doses: 500 mg daily in combination with one or more other agents for treatment, or 1200 mg once weekly for primary prevention. The treatment of uncomplicated nongonococcal urethritis presumed to be due to C. trachomatis consists of a single 1-g dose of azithromycin. A single 2-g dose is effective for gonorrhea, but is not routinely recommended (Centers for Disease Control and Prevention, 1998). In children, the recommended dose of oral suspension for acute otitis media and pneumonia is 10 mg/kg the first day (maximum 500 mg) and 5 mg/kg (maximum 250 mg per day) on days 2 through 5. The dose for tonsillitis or pharyngitis is 12 mg/kg per day, up to 500 mg total, for 5 days. Mycoplasma pneumoniae Infections Erythromycin (given orally in doses of 500 mg four times daily, or, if oral administration is not tolerated, given intravenously) reduces the duration of fever caused by M. pneumoniae. In addition, the rate of clearing, as indicated by chest radiographs, is accelerated (Rasch and Mogabgab, 1965). Tetracycline or another macrolide is just as effective. Legionnaires' Disease Erythromycin has been considered as the drug of choice for treatment of pneumonia caused by L. pneumophila, L. micdadei, or other Legionella spp. Azithromycin has supplanted erythromycin as the first-line agent (along with fluoroquinlones) for treatment of legionellosis because of excellent in vitro activity, superior tissue concentration, the ease of administration as a single daily dose, and better tolerability compared to erythromycin (Stout et al., 1998; Garey and Amsden, 1999; Yu, 2000). The recommended dose is 500 mg intravenously or orally for a total of 10 to 14 days. Chlamydia Infections Chlamydial infections can be treated effectively with any of the macrolides. Azithromycin is specifically recommended as an alternative to doxycycline in patients with uncomplicated urethral, endocervical, rectal, or epididymal infections (Centers for Disease Control and Prevention, 1998). Clearly the major impact of azithromycin is due to the better compliance that results from a single-dose treatment regimen. During pregnancy, erythromycin base, 500 mg four times daily for 7 days, is recommended as first-line therapy for chlamydial urogenital infections. Azithromycin, 1 g orally as a single dose, is a suitable alternative (Centers for Disease Control and Prevention, 1998). Erythromycin base is preferred for chlamydial pneumonia of infancy and ophthalmia neonatorum (50 mg/kg per day in four divided doses for 10 to 14 days), as tetracyclines are contraindicated in this patient group. Pneumonia caused by Chlamydia pneumoniae responds to macrolides, fluoroquinolones, and tetracyclines in standard doses for community-acquired pneumonia. No comparative trials have been conducted to determine which, if any, agent is most efficacious. Duration of therapy also is ill defined. A two-week duration of therapy has been recommended (Bartlett et al., 1993), although in practice a specific etiological diagnosis rarely is made and length of treatment often is determined empirically based on clinical response. Diphtheria Erythromycin is very effective for acute infections or for eradicating the carrier state. Erythromycin estolate (250 mg four times daily for 7 days) was found to be effective in 90% of adults. The other macrolides also are likely to be effective, but clinical experience with them is lacking, and they are not FDA-approved for this indication. Neither erythromycin nor any other antibiotic alters the course of an acute infection with the diphtheria bacillus or the risk of complications. Antitoxin is indicated in the treatment of acute infection. Pertussis Erythromycin is the drug of choice for treating persons with B. pertussis disease and for post-exposure prophylaxis of all household members and other close contacts. A 7-day regimen of erythromycin estolate (40 mg/kg per day, maximum 1 g/day) is as effective as 14-day erythromycin regimens traditionally recommended (Halperin et al., 1997). Clarithromycin and azithromycin appear to be just as effective, although clinical experience is limited (Aoyama et al., 1996; Bace et al., 1999). If administered early in the course of whooping cough, erythromycin may shorten the duration of illness. The drug has little influence on the disease once the paroxysmal stage is reached, although it may eliminate the microorganisms from the nasopharynx. Nasopharyngeal cultures should be obtained from persons with pertussis who do not improve with erythromycin therapy, as resistance has been reported (Centers for Disease Control, 1994). Streptococcal Infections Pharyngitis, scarlet fever, erysipelas, and cellulitis caused by S. pyogenes and pneumonia caused by S. pneumoniae respond to macrolides. They are valuable alternatives for treatment of patients who have serious allergy to penicillin. Unfortunately, macrolide-resistant strains are increasingly encountered and may cause infections that do not respond to these agents. As noted above, penicillin-resistant strains of S. pneumoniae are very likely also to be resistant to macrolides. Staphylococcal Infections Erythromycin has been an alternative agent for the treatment of relatively minor infections caused by either penicillin-sensitive or penicillin-resistant S. aureus. However, many strains of S. aureus, including community-acquired isolates, are resistant to macrolides, such that these agents no longer can be relied upon unless in vitro susceptibility has been documented. Campylobacter Infections The treatment of gastroenteritis caused by Campylobacter jejuni with erythromycin (250 to 500 mg orally four times a day for 7 days) hastens eradication of the microorganism from the stools and reduces the duration of symptoms (Salazar-Lindo et al., 1986). Availability of fluoroquinolones, which are highly active against Campylobacter species and other enteric pathogens, has largely replaced the need for erythromycin for this disease in adults. Erythromycin remains useful for treatment of Campylobacter gastroenteritis in children. Helicobacter pylori Infection Clarithromycin 500 mg, in combination with omeprazole, 20 mg, and amoxicillin, 1g, each administered twice daily for 10 to 14 days is effective for treatment of peptic ulcer disease caused by H. pylori (Peterson et al., 2000). Numerous other regimens, some effective as a seven-day treatment, have been studied and also are effective (Misiewicz et al., 1997; Hunt et al., 1999). The more effective regimens generally include three agents, one of which usually is clarithromycin. Tetanus Erythromycin (500 mg orally every 6 hours for 10 days) may be given to eradicate Clostridium tetani in patients with tetanus who are allergic to penicillin. However, the mainstays of therapy are debridement, physiological support, tetanus antitoxin, and drug control of convulsions. Syphilis Erythromycin has been used in the treatment of early syphilis in patients who are allergic to penicillin, but is no longer recommended (Centers for Disease Control and Prevention, 1998). Tetracyclines are the recommended alternative in penicillin-allergic patients. During pregnancy it is recommended that patients be desensitized to penicillin. Mycobacterial Infections Clarithromycin or azithromycin is recommended as first-line therapy for prophylaxis and treatment of disseminated infection caused by M. avium-intracellulare in AIDS patients and for treatment of pulmonary disease in non-HIV-infected patients (American Thoracic Society 1997; Kovacs and Masur 2000). Azithromycin (1200 mg once weekly) or clarithromycin (500 mg twice daily) is recommended for primary prevention for AIDS patients with less than 50 CD4 cells per mm3. Single-agent therapy should not be used for treatment of active disease or for secondary prevention in AIDS patients. First-line therapy is clarithromycin (500 mg twice daily) plus ethambutol (15 mg/kg once daily) with or without rifabutin. Azithromycin (500 mg once daily) may be used instead of clarithromycin, but clarithromycin appears to be slightly more efficacious (Ward et al., 1998). Clarithromycin also has been used with minocycline for the treatment of Mycobacterium leprae in lepromatous leprosy (Ji et al., 1993). Other Infections Clarithromycin and azithromycin have been used in the treatment of toxoplasmosis encephalitis (Saba et al., 1993) and diarrhea due to Cryptosporidium (Rehg, 1991) in AIDS patients. Rigorous clinical trials demonstrating efficacy of macrolides for these infections are lacking. Prophylactic Uses Penicillin is the drug of choice for the prophylaxis of recurrences of rheumatic fever, but it cannot be used in individuals who are allergic to this antibiotic. Erythromycin is an effective alternative. Erythromycin has been recommended as an alternative to penicillin in allergic patients for prevention of bacterial endocarditis following dental or respiratory-tract procedures (Dajani et al., 1990). Clindamycin has replaced erythromycin for use in penicillin-allergic patients. Clarithromycin or azithromycin as a single 500-mg dose also may be used (Dajani et al., 1997). Untoward Effects Serious untoward effects are only rarely caused by erythromycin. Among the allergic reactions observed are fever, eosinophilia, and skin eruptions, which may occur alone or in combination; each disappears shortly after therapy is stopped. Cholestatic hepatitis is the most striking side effect. It is caused primarily by erythromycin estolate and only rarely by the ethylsuccinate or the stearate (seeGinsburg and Eichenwald, 1976). The illness starts after about 10 to 20 days of treatment and is characterized initially by nausea, vomiting, and abdominal cramps. The pain often mimics that of acute cholecystitis, and unnecessary surgery has been performed. These symptoms are followed shortly thereafter by jaundice, which may be accompanied by fever, leukocytosis, eosinophilia, and elevated activities of transaminases in plasma. Biopsy of the liver reveals cholestasis, periportal infiltration by neutrophils, lymphocytes, and eosinophils, and, occasionally, necrosis of neighboring parenchymal cells. All manifestations usually disappear within a few days after cessation of drug therapy and rarely are prolonged. The syndrome may represent a hypersensitivity reaction to the estolate ester (seeTolman et al., 1974). Mild elevations of serum aspartate aminotransferase enzymes also may occur (McCormack et al., 1977). Oral administration of erythromycin, especially of large doses, frequently is accompanied by epigastric distress, which may be quite severe. Intravenous administration of erythromycin may cause similar symptoms, with abdominal cramps, nausea, vomiting, and diarrhea. Erythromycin may stimulate gastrointestinal motility by acting on motilin receptors (Smith et al., 2000). The gastrointestinal symptoms are dose-related and occur more commonly in children and young adults (Seifert et al., 1989); they may be reduced by prolonging the infusion time to 1 hour or by pretreatment with glycopyrrolate (Bowler et al., 1992). Intravenous infusion of 1-g doses, even when dissolved in a large volume, often is followed by thrombophlebitis. This can be minimized by slow rates of infusion. Erythromycin has been reported to cause cardiac arrhythmias, including QT prolongation with ventricular tachycardia. Most patients have had underlying cardiac disease, or the arrhythmias were associated with combination drug therapies that included erythromycin (e.g., cisapride or terfenadine plus erythromycin) (Brandriss et al., 1994). Transient auditory impairment is a potential complication of treatment with erythromycin; it has been observed to follow intravenous administration of large doses of the gluceptate or lactobionate (4 g per day) or oral ingestion of large doses of the estolate (Karmody and Weinstein, 1977). Drug Interactions Erythromycin and clarithromycin have been reported to cause clinically significant drug interactions (Periti et al., 1992). Erythromycin has been reported to potentiate the effects of astemizole, carbamazapine, corticosteroids, cyclosporine, digoxin, ergot alkaloids, terfenadine, theophylline, triazolam, valproate, and warfarin, probably by interfering with cytochrome P450-mediated metabolism of these drugs (Ludden, 1985; Martell et al., 1986; Honig et al., 1992). Clarithromycin, which is structurally closely related to erythromycin, has a similar drug interaction profile. Azithromycin, which differs from erythromycin and clarithromycin because of its 15-membered lactone ring structure, appears to be free of these drug interactions. Caution is advised, nevertheless, when using azithromycin in conjuction with drugs known to interact with erythromycin. |
Clindamycin
|
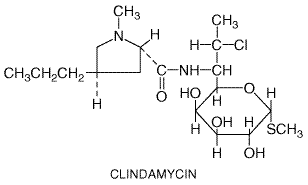
Chemistry Clindamycin is a derivative of the amino acid trans-L-4-n-propylhygrinic acid, attached to a sulfur-containing derivative of an octose. It is a congener of lincomycin, and its structural formula is as follows:
Mechanism of Action Clindamycin binds exclusively to the 50 S subunit of bacterial ribosomes and suppresses protein synthesis. Although clindamycin, erythromycin, and chloramphenicol are not structurally related, they all act at sites within close proximity (seeFigures 472 and 473), and binding by one of these antibiotics to the ribosome may inhibit the interaction of the others. There are no clinical indications for the concurrent use of these antibiotics. Macrolide resistance due to ribosomal methylation by erm-encoded enzymes also may produce resistance to clindamycin. However, because clindamycin is not an inducer of the methylase, there is cross-resistance only if the enzyme is produced constitutively. Clindamycin is not a substrate for macrolide efflux pumps, and strains that are resistant to macrolides by this mechanism are susceptible to clindamycin. Plasmid-mediated resistance to clindamycin (and erythromycin) has been found in B. fragilis (Tally et al., 1979); it may be due to methylation of bacterial RNA found in the 50 S ribosomal subunit (Steigbigel, 2000). Antibacterial Activity Bacterial strains are susceptible to clindamycin at minimal inhibitory
concentrations of Clindamycin is more active than erythromycin or clarithromycin against
anaerobic bacteria, especially B. fragilis; some strains are inhibited
by <0.1 With regard to atypical organisms and parasites, M. pneumoniae is resistant. Chlamydia spp. are variably sensitive, although the clinical relevance is not established. Clindamycin shows good activity in experimental models of Pneumocystis carinii pneumonia and T. gondii encephalitis. Clindamycin has some activity against both chloroquine-sensitive and chloroquine-resistant strains of Plasmodium falciparum and Plasmodium vivax, but a cure rate of only 50% of patients with malaria was observed in one study (Hall et al., 1975; see also Seaberg et al., 1984). Clindamycin has been used for treatment of babesiosis. Absorption, Distribution, and Excretion Absorption Clindamycin is nearly completely absorbed following oral
administration. Peak plasma concentrations of 2 to 3 Clindamycin palmitate, an oral preparation for pediatric use, is an
inactive prodrug, but the ester is hydrolyzed rapidly in vivo. Its
rate and extent of absorption are similar to those of clindamycin. After
several oral doses at 6-hour intervals, children attain plasma concentrations
of 2 to 4 The phosphate ester of clindamycin, which is given parenterally, also
is rapidly hydrolyzed in vivo to the active parent compound. After
intramuscular injection, peak concentrations in plasma are not attained until
3 hours in adults and 1 hour in children; these values approximate 6 Distribution Clindamycin is widely distributed in many fluids and tissues, including bone. Significant concentrations are not attained in CSF, even when the meninges are inflamed. Concentrations sufficient to treat cerebral toxoplasmosis are achievable (Gatti et al., 1998). The drug readily crosses the placental barrier. Ninety percent or more of clindamycin is bound to plasma proteins (seePanzer et al., 1972). Clindamycin accumulates in polymorphonuclear leukocytes, alveolar macrophages, and in abscesses. Excretion Only about 10% of the clindamycin administered is excreted unaltered in the urine, and small quantities are found in the feces. However, antimicrobial activity persists in feces for 5 or more days after parenteral therapy with clindamycin is stopped; growth of clindamycin-sensitive microorganisms in colonic contents may remain suppressed for up to 2 weeks (Kager et al., 1981). Clindamycin is inactivated by metabolism to N-demethylclindamycin and clindamycin sulfoxide, which are excreted in the urine and bile. Accumulation of clindamycin can occur in patients with severe hepatic failure, and dosage adjustments thus may be required. Therapeutic Uses The oral dose of clindamycin (clindamycin hydrochloride; CLEOCIN ) for adults is 150 to 300 mg every 6 hours; for severe infections, it is 300 to 600 mg every 6 hours. Children should receive 8 to 12 mg/kg per day of clindamycin palmitate hydrochloride (CLEOCIN PEDIATRIC) in three or four divided doses (some physicians recommend 10 to 30 mg/kg per day in six divided doses) or for severe infections, 13 to 25 mg/kg per day. However, children weighing 10 kg or less should receive 1/2 teaspoonful of clindamycin palmitate hydrochloride (37.5 mg) every 8 hours as a minimal dose. For serious infections due to aerobic gram-positive cocci and the more sensitive anaerobes (not generally including B. fragilis, Peptococcus, and Clostridium spp. other than C. perfringens), intravenous or intramuscular administration is recommended in dosages of 600 to 1200 mg per day, divided into two to four equal portions for adults. Clindamycin phosphate ( CLEOCIN PHOSPHATE) is available for intramuscular or intravenous use. For more severe infections, particularly those proven or suspected to be caused by B. fragilis, Peptococcus, or Clostridium species other than C. perfringens, parenteral administration of 1200 to 2400 mg per day of clindamycin is suggested. Daily doses as high as 4800 mg have been given intravenously to adults. Children should receive 10 to 40 mg/kg per day in three or four divided doses. In severe infections, a minimal daily dose of 300 mg is recommended, regardless of body weight. Although a number of infections with gram-positive cocci will respond
favorably to clindamycin, the high incidence of diarrhea and the occurrence
of colitis require limitation of its use to infections in which it is clearly
superior to other agents. Clindamycin is particularly valuable for the
treatment of infections with anaerobes, especially those due to B.
fragilis. It has been used successfully in combination with an
aminoglycoside for infections resulting from fecal spillage (intraabdominal
or pelvic abscesses and peritonitis). Other drugs that are effective against
anaerobes, such as metronidazole, chloramphenicol, cefoxitin, cefmetazole, cefotetan,
ceftizoxime, cefotaxime, imipenem, or penicillins plus One randomized, prospective trial has shown that clindamycin (600 mg
intravenously every 8 hours) was superior to penicillin (1 million units
intravenously every 4 hours) for the treatment of lung abscesses (Levison et
al., 1983). On the basis of this study, the results of which continue to
be debated ( Clindamycin (600 to 1200 mg given intravenously every 6 hours) in combination with pyrimethamine (a 200-mg loading dose followed by 75 mg orally each day) and folinic acid (10 mg/day) has been shown to be effective for acute treatment of encephalitis caused by T. gondii in patients with AIDS (Dannemann et al., 1992; Katlama et al., 1996). Clindamycin (600 mg intravenously every 8 hours, or 300 to 450 orally every 6 hours for less severe disease) in combination with primaquine (15 mg of base once daily) has been shown to be useful for the treatment of mild to moderate cases of P. carinii pneumonia (PCP) in AIDS patients (Black et al., 1994; Toma et al., 1993). Clindamycin also is available as a topical solution, gel, or lotion ( CLEOCIN T , others) and as a vaginal cream ( CLEOCIN ). It is effective topically (or orally) for acne vulgaris and bacterial vaginosis. Untoward Effects The reported incidence of diarrhea associated with the administration of clindamycin ranges from 2% to 20%. A number of patients (variously reported as 0.01% to 10%) have developed pseudomembranous colitis caused by the toxin from the organism C. difficile (Rifkin et al., 1977). This colitis is characterized by abdominal pain, diarrhea, fever, and mucus and blood in the stools. Proctoscopic examination reveals white to yellow plaques on the mucosa of the colon. This syndrome may be lethal. Discontinuation of the drug, combined with administration of metronidazole orally or intravenously usually is curative, but relapses occur. Agents that inhibit peristalsis, such as opioids, may prolong and worsen the condition. Although the incidence of this problem is unknown, it is clear that the therapeutic indications for clindamycin, or any antibiotic, should be considered very seriously before it is given. Skin rashes occur in approximately 10% of patients treated with clindamycin and may be more common in patients with human immunodeficiency virus (HIV) infection. Other reactions, which are uncommon, include exudative erythema multiforme (Stevens-Johnson syndrome), reversible elevation of aspartate aminotransferase and alanine aminotransferase, granulocytopenia, thrombocytopenia, and anaphylactic reactions. Local thrombophlebitis may follow intravenous administration of the drug. Clindamycin can inhibit neuromuscular transmission and may potentiate the effect of a neuromuscular blocking agent administered concurrently (Fogdall and Miller, 1974). |
Quinupristin/Dalfopristin
|
Quinupristin/dalfopristin SYNECID) is a combination of a
streptogramin B, quinupristin, with a streptogramin A, dalfopristin, in a
30:70 ratio (Chant and Rybak 1995). These compounds are semisynthetic
derivatives of naturally occurring pristinamycins, produced by Streptomyces
pristinaespiralis. Pristinamycin has been available as an oral agent in
Antibacterial Activity Quinupristin/dalfopristin is active against gram-positive cocci,
including S. pneumoniae, beta-hemolytic and alpha-hemolytic strains of
streptococci, E. faecium (but not E. faecalis), and
staphylococci, both coagulase-positive and coagulase-negative strains (Chang et
al., 1999). The combination is largely inactive against gram-negative
organisms, although Moraxella catarrhalis and Neiserria spp.
are susceptible. It is also active against organisms responsible for atypical
pneumonia, M. pneumoniae, Legionella spp., and Chlamydia pneumoniae
(Lamb et al., 1999). The combination is bactericidal against
streptococci and many strains of staphylococci, but bacteriostatic against E.
faecium. MICs for strains of streptococci, including
penicillin-susceptible and penicillin-resistant strains of S. pneumoniae,
are 0.25 to 1 Mechanism of Action Quinupristin and dalfopristin are protein synthesis inhibitors that bind the 50 S ribosomal subunit. Quinupristin, a type B streptogramin, binds at the same site as macrolides and has a similar effect, with inhibition of polypeptide elongation and early termination of protein synthesis. Dalfopristin binds at a site nearby, resulting in a conformational change in the 50 S ribosome, synergistically enhancing the binding of quinupristin at its target site. Dalfopristin directly interferes with polypeptide-chain formation. The net effect, in many bacterial species, of the cooperative and synergistic binding of these two molecules to the ribosome is bactericidal activity. Resistance to quinupristin is mediated by MLS type B resistance determinants (e.g., ermA and ermC in staphylococci and ermB in enterococci), encoding a ribosomal methylase that prevents binding of drug to its target; or vgb or vgbB, which encode lactonases that inactivate type B streptogramins (Allignet et al., 1998; Bozdogan and Leclercq, 1999). Resistance to dalfopristin is mediated by vat, vatB, vatC, vatD, and satA, which encode acetyltransferases that inactivate type A streptogramins (Allignet et al., 1998; Allignet and El Solh, 1999; Soltani et al., 2000); or staphylococcal genes vga, vgb, and vgaB, which encode ATP-binding efflux proteins that pump type A compounds out of the cell (Allignet et al., 1998; Bozdogan and Leclercq, 1999). These resistance determinants are located on plasmids that may be transferable by conjugative mobilization (Allignet et al., 1998). Resistance to quinupristin/dalfopristin always is associated with a resistance gene for type A streptogramins. Genes encoding resistance to type B streptogramins also may be present, but are not sufficient to produce resistance alone. Methylase-encoding erm genes, however, can render the combination bacteriostatic instead of bactericidal, rendering it ineffective in certain infections in which bactericidal activity is necessary for cure, such as endocarditis (Chambers, 1992; Fantin et al., 1995). Absorption, Distribution, and Excretion Quinupristin/dalfopristin is administered only by intravenous infusion
over at least one hour. It is incompatible with saline and heparin and should
be dissolved in 5% dextrose in water. Steady-state peak serum concentrations
in healthy male volunteers are approximately 3 Therapeutic Uses Quinupristin/dalfopristin is approved in the Untoward Effects The most common side effects are infusion-related events, such as pain and phlebitis at the infusion site and arthralgias and myalgias. Phlebitis and pain can be minimized by infusion of drug through a central venous catheter. Arthalgias and myalgias, which are more likely to be a problem in patients with hepatic insufficiency and may be due to accumulation of metabolites, is managed by reducing the infusion frequency to every 12 hours. Quinupristin/dalfopristin is an inhibitor of cytochrome P450 enzyme 3A4 (CYP3A4). Drugs that are metabolized by CYP3A4 include terfenadine, astemizole, indinavir, nevirapine, midazolam, nifedipine and other calcium channel blockers, and cyclosporine. Concomitant administration of quinupristin/dalfopristin with these or other drugs metabolized by CYP3A4 may enhance drug effects and result in significant toxicity. Appropriate caution and monitoring are recommended for drugs in which the toxic therapeutic window is narrow or for drugs that cause QTc prolongation (e.g., some antihistamines). |
Linezolid
|
Linezolid ZYVOX) is a synthetic antimicrobial
agent of the oxazolidinone class (Zurenko et al., 1996; Diekema and
Jones, 2000). Its chemical structure is: Antibacterial Activity Linezolid is active against grampositive organisms including
staphylococci, streptococci, enterococci, gram-positive anaerobic cocci, and
gram-positive rods such as Corynebacterium spp. and Listeria
monocytogenes (Jones et al., 1996; Zurenko et al., 1996).
It has poor activity against most gram-negative aerobic or anaerobic
bacteria. It is bacteriostatic against enterococci and staphylococci and
bactericidal against streptococci. MICs are Mechanism of Action Linezolid inhibits protein synthesis. Linezolid prevents formation of the 70 S ribosome complex that initiates protein synthesis by binding to the 23 S subunit of the 50 S subunit. Because of its unique binding site and its action at the early, ribosome-assembly step of protein synthesis, there is no cross-resistance with other drug classes. Resistance is due to mutation of the ribosomal binding site (Kloss et al., 1999; Hamel et al., 2000). Resistance has been reported clinically only for enterococci, although resistant mutants have been selected from strains of S. aureus by passage in linezolid in vitro. Absorption, Distribution, and Excretion Linezolid is well absorbed after oral administration. It may be
administered without regard to food. With oral bioavailability approaching
100%, dosing for oral and intravenous preparations is the same. Peak serum
concentrations average 12 to 14 Linezolid is metabolized by nonenzymatic oxidation to aminoethoxyacetic acid and hydroxyethyl glycine metabolites. Approximately 80% of the dose of linezolid appears in the urine, 30% as active compound, and 50% as the two primary metabolites. Ten percent of the administered dose appears as metabolites in feces. Serum concentrations and half-life of the parent compound are not appreciably altered by renal insufficiency. The metabolites accumulate in renal insufficiency, with half-lives increasing by approximately 50% to 100%. The clinical significance of this is unknown, and no dose adjustment is recommended at this time. Linezolid and its metabolites are eliminated by dialysis; therefore, the drug should be administered following hemodialysis. No data concerning the effect of peritoneal dialysis are available. Therapeutic Uses Linezolid is approved by the United States Food and Drug Administration for treatment of infections caused by vancomycin-resistant E. faecium; nosocomial pneumonia caused by methicillin-susceptible and -resistant strains of S. aureus; community-acquired pneumonia caused by penicillin-susceptible strains of S. pneumoniae; complicated skin and skin-structure infections caused by streptococci and methicillin-susceptible and -resistant strains of S. aureus; and uncomplicated skin and skin-structure infections (Clemett and Markham, 2000). In noncomparative studies, linezolid (600 mg twice daily) has had clinical and microbiological cure rates in the range of 85% to 90% in treatment of a variety of infections (soft tissue, urinary tract and bacteremia) caused by vancomycin-resistant E. faecium. A 200-mg twice-daily dose was less effective, with a clinical cure rate of approximately 75% and a microbiological cure rate of only 59%. The 600-mg twice-daily dose and not the 200-mg dose, therefore, should be used for treatment of infections caused by enterococci. A 400-mg twice-daily dosage regimen is recommended only for treatment of uncomplicated skin and skin-structure infections. In randomized, comparative studies, cures rates with linezolid (which were about 60%) were similar to those with vancomycin for nosocomial pneumonia caused by methicillin-resistant or -susceptible S. aureus. Efficacy of linezolid also was similar to that of either oxacillin or vancomycin for skin and skin-structure infections, the majority of microbiologically documented cases being caused by S. aureus. Although relatively few patients with S. aureus bacteremia have been treated, linezolid appears to be comparable in efficacy to vancomycin for methicillin-resistant strains. However, linezolid is bacteriostatic for staphylococci and enterococci, and it should not be used for treatment of suspected endocarditis. Linezolid should be reserved as an agent of last resort for treatment of infections caused by multiple-drug-resistant strains. It should not be used when alternative agents are likely to be effective (e.g., community-acquired pneumonia, even though it has the indication). Indiscriminant use and overuse will hasten selection of resistant strains and the eventual loss of this valuable new agent. Untoward Effects Data on toxicity and side effects are extremely limited at present. The drug seems to be well tolerated, with generally minor side effects (e.g., gastrointestinal complaints, headache, rash) reported at rates no different from comparator agents in clinical trials. Thrombocytopenia or a significant reduction in platelet count has been associated with linezolid. The reported incidence is 2.4%, and its occurrence is related to duration of therapy. Platelet counts should be monitored in patients with risk of bleeding, preexisting thrombocytopenia, or intrinsic or acquired disorders of platelet function (including those potentially caused by concomitant medication) and in patients receiving courses of therapy lasting beyond two weeks. Linezolid is a weak, nonspecific monoamine-oxidase inhibitor. Patients receiving concomitant therapy with an adrenergic or serotonergic agent or consuming more than 100 mg of tyramine a day may experience an enhancement of drug effect. No other significant drug interactions have been identified. Specifically, linezolid is neither a substrate nor an inhibitor of cytochrome P450 enzymes. |
Spectinomycin
|
Source and Chemistry Spectinomycin is an antibiotic produced by Streptomyces spectabilis. The drug is an aminocyclitol; its structural formula is as follows:
Antibacterial Activity and Mechanism Spectinomycin is active against a number of gram-negative bacterial species, but it is inferior to other drugs to which such microorganisms are susceptible (Schoutens et al., 1972). Its only therapeutic use is in the treatment of gonorrhea caused by strains resistant to first-line drugs or if there are contraindications to the use of these drugs. Resistance, although rare, does occur (Clendennen et al., 1992). Spectinomycin selectively inhibits protein synthesis in gram-negative bacteria. The antibiotic binds to and acts on the 30 S ribosomal subunit. Its action is similar to that of the aminoglycosides; however, spectinomycin is not bactericidal and does not cause misreading of messenger RNA. Bacterial resistance may develop as a result of mutation or a modifying enzyme (Clark et al., 1999). Absorption, Distribution, and Excretion Spectinomycin is rapidly absorbed after intramuscular injection. A
single dose of 2 g produces peak serum concentrations of 100 Therapeutic Uses The Centers for Disease Control and Prevention recommends ceftriaxone,
cefixime, ciprofloxacin, or ofloxacin for the treatment of uncomplicated
gonococcal infection. However, spectinomycin is recommended as an alternative
regimen in patients who are intolerant or allergic to Untoward Effects Spectinomycin, when given as a single intramuscular injection, produces few significant untoward effects (Duncan et al., 1972). Urticaria, chills, and fever have been noted after single doses, as have dizziness, nausea, and insomnia. The injection may be painful. |
Polymyxin B and Colistin
|
Because of the extreme nephrotoxicity associated with parenteral administration of these drugs, they are now rarely if ever used except topically. Source and Chemistry The polymyxins, discovered in 1947, are a group of closely
related antibiotic substances elaborated by various strains of Bacillus
polymyxa, an aerobic spore-forming rod found in soil. Colistin (polymyxin
E) is produced by Bacillus (Aerobacillus) colistinus, a microorganism
isolated from a soil sample obtained from Colistin is polymyxin E, and it has a similar structure; it is available for clinical use as colistin sulfate, for oral use, and as colistimethate sodium, a parenteral preparation. Antibacterial Activity and Mechanism of Action The antimicrobial activities of polymyxin B and colistin are similar
and are restricted to gram-negative bacteria, including Enterobacter, E.
coli, Klebsiella, Salmonella, Pasteurella, Bordetella, and Shigella,
which usually are sensitive to concentrations of 0.05 to 2.0 Polymyxins are surface-active, amphipathic agents (containing both lipophilic and lipophobic groups within the molecule). They interact strongly with phospholipids and penetrate into and disrupt the structure of cell membranes. The permeability of the bacterial membrane changes immediately on contact with the drug. Sensitivity to polymyxin B apparently is related to the phospholipid content of the cell wallmembrane complex (Brown and Wood, 1972). The cell wall of certain resistant bacteria may prevent access of the drug to the cell membrane. Polymyxin B binds to the lipid A portion of endotoxin (the lipopolysaccharide of the outer membrane of gram-negative bacteria) and inactivates this molecule. Polymyxin B attenuates pathophysiologic consequences of the release of endotoxin in several experimental systems (Shenep et al., 1984; Tauber et al., 1987). The clinical utility of polymixin B for this indication has not yet been established. Absorption, Distribution, and Excretion Neither polymyxin B nor colistin is absorbed when given orally. They are also poorly absorbed from mucous membranes and the surface of large burns. Therapeutic Uses Polymyxin B sulfate is available for ophthalmic, otic, and topical use in combination with a variety of other compounds. Although parenteral preparations are still marketed, they are not recommended. Infections of the skin, mucous membranes, eye, and ear due to polymyxin B-sensitive microorganisms respond to local application of the antibiotic in solution or ointment. External otitis, frequently due to Pseudomonas, may be cured by the topical use of the drug. P. aeruginosa is a common cause of infection of corneal ulcers; local application or subconjunctival injection of polymyxin B often is curative. Untoward Effects Polymyxin B applied to intact or denuded skin or mucous membranes produces no systemic reactions because of its almost complete lack of absorption from these sites. Hypersensitization is uncommon with topical application. Adverse effects that follow the parenteral administration of these drugs are discussed in the fifth edition of this textbook. |
Vancomycin
|
History and Source Vancomycin is an antibiotic produced by Streptococcus orientalis,
an actinomycete isolated from soil samples obtained in Chemistry Vancomycin is a complex and unusual tricyclic glycopeptide with a molecular mass of about 1500 daltons. Its structural formula was determined by x-ray analysis (Sheldrick et al., 1978), and is as follows:
Antibacterial Activity Vancomycin is primarily active against gram-positive bacteria. Strains
are considered susceptible at MICs of S. pyogenes, S. pneumoniae, and viridans streptococci are highly
susceptible to vancomycin. Corynebacterium spp. (diphtheroids) are
inhibited by less than 0.04 to 3.1 Mechanisms of Action and Resistance Vancomycin inhibits the synthesis of the cell wall in sensitive bacteria by binding with high affinity to the D-alanyl-D-alanine terminus of cell wall precursor units (seeFigure 474). The drug is bactericidal for dividing microorganisms.
Enterococcal resistance to vancomycin is the result of alteration of the D-alanyl-D-alanine target to D-alanyl-D-lactate or D-alanyl-D-serine (Walsh, 1993; Arias et al., 2000), which bind vancomycin poorly, because a critical site for hydrogen bonding is missing. Several enzymes within the van gene cluster are required for this target alteration to occur. Several phenotypes of resistance to vancomycin have been described. The Van A phenotype confers resistance to both teicoplanin and vancomycin. The trait is inducible and has been identified in E. faecium and E. faecalis. The Van B phenotype, which tends to be a lower level of resistance, also has been identified in E. faecium and E. faecalis. The trait is inducible by vancomycin but not teicoplanin, and, consequently, many strains remain susceptible to teicoplanin. The Van C phenotype, the least important clinically and least well characterized, confers resistance only to vancomycin, is constitutive, and is present in no species of enterococci other than E. faecalis and E. faecium. Van D and Van E gene clusters also have been identified and others presumably will follow. The genetic and biochemical basis of reduced susceptibility to vancomycin in Staphylococcus is not well understood. Several genetic elements and multiple mutations are required. Many of the genes that have been implicated encode enzymes of the cell-wall biosynthetic pathway (Hanaki et al., 1998; Sieradzki and Tomasz 1999; Sieradzki et al., 1999b). Absorption, Distribution, and Excretion Vancomycin is poorly absorbed after oral administration, and large
quantities are excreted in the stool. For parenteral therapy, the drug should
be administered intravenously, never intramuscularly. A single intravenous
dose of 1 g in adults produces plasma concentrations of 15 to 30 Therapeutic Uses Vancomycin hydrochloride VANCOCIN ,
others) is marketed for intravenous use as a sterile powder for
solution. It should be diluted and infused over at least a 60-minute period
to avoid infusion-related adverse reactions (see below). The dose of
vancomycin for adults is 30 mg/kg per day in 2 to 4 divided doses. A higher
dose, 60 mg/kg per day in 4 divided doses, is recommended for meningitis (Quagliarello
and Scheld, 1997). This regimen will yield an average steady-state
concentration of 15 Pediatric doses are as follows: for newborns during the first week of life, 15 mg/kg initially, followed by 10 mg/kg every 12 hours; for infants 8 to 30 days old, 15 mg/kg followed by 10 mg/kg every 8 hours; for older infants and children, 10 mg/kg every 6 hours (Schaad et al., 1980). Alteration of dosage is required for patients with impaired renal function (seeAppendix II). The drug has been used effectively in functionally anephric patients (who are being dialyzed) by the administration of 1 g (approximately 15 mg/kg) each week. Vancomycin can be administered orally to patients with pseudomembranous colitis, although metronidazole is preferred. The dose for adults is 125 to 250 mg every 6 hours; the total daily dose for children is 40 mg/kg, given in three to four divided doses. Vancomycin hydrochloride for oral solution is available for this purpose, as are capsules. Vancomycin should be employed only to treat serious infections and is
particularly useful in the management of infections due to
methicillin-resistant staphylococci, including pneumonia, empyema,
endocarditis, osteomyelitis, and soft-tissue abscesses and in severe
staphylococcal infections in patients who are allergic to penicillins and
cephalosporins (Geraci, 1977). However, vancomycin is less rapidly
bactericidal than any of the antistaphylococcal Administration of vancomycin is an effective alternative for the treatment of endocarditis caused by viridans streptococci in patients who are allergic to penicillin. In combination with an aminoglycoside, it may be used for enterococcal endocarditis in patients with serious penicillin allergy. Vancomycin also is effective for the treatment of infections caused by Flavobacterium and Corynebacterium spp. Vancomycin has become an important antibiotic in the management of known or suspected penicillin-resistant pneumococcal infections (Friedland and McCracken, 1994). Untoward Effects Among the hypersensitivity reactions produced by vancomycin are macular skin rashes and anaphylaxis. Phlebitis and pain at the site of intravenous injection are relatively uncommon. Chills, rash, and fever may occur. Rapid intravenous infusion may cause a variety of symptoms, including erythematous or urticarial reactions, flushing, tachycardia, and hypotension. The extreme flushing that can occur is sometimes called 'red-neck' or 'red-man' syndrome (Newfield and Roizen, 1979; Davis et al., 1986). Auditory impairment, which is frequently although not always
permanent, may follow the use of this drug. Ototoxicity is associated with
excessively high concentrations of the drug in plasma (60 to 100 |
Teicoplanin
|
Source and Chemistry Teicoplanin is a glycopeptide antibiotic produced by Actinoplanes
teichomyetius. The drug actually is a mixture of six closely related
compounds: one compound has a terminal hydrogen at the oxygen indicated by an
asterisk; five compounds have an R substituent of either a decanoic acid [n-,
8-methyl-, 9-methyl, (Z)-4-] or of a nonanoic acid [8-methyl].
Although not FDA-approved for use in the
Mechanisms of Action and Resistance Teicoplanin, like vancomycin, is an inhibitor of cell-wall synthesis,
and it is active only against gram-positive bacteria. It is reliably
bactericidal against susceptible strains, except for enterococci. It is
active against methicillin-susceptible and methicillin-resistant
staphylococci, which typically have minimal inhibitory concentrations of
<4 The mechanisms of resistance to teicoplanin in strains of staphylococci have not been elucidated, but resistance can emerge in a previously susceptible strain during a course of therapy (Kaatz et al., 1990). The Van A phenotype of vancomycin-resistant enterococci also determines resistance to teicoplanin. The mechanism is the same as for vancomycin: alteration of the cell wall target such that the glycopeptide does not bind. Strains of enterococci with Van B resistance often are susceptible to teicoplanin, because it is a poor inducer of the enzymes responsible for the cell wall alteration. Van C strains of enterococci, which in general are not human pathogens, are susceptible to teicoplanin (Arthur and Courvalin, 1993). Absorption, Distribution, and Excretion The primary differences between vancomycin and teicoplanin are that
teicoplanin can be administered safely by intramuscular injection; it is
highly bound by plasma proteins (90% to 95%); and it has an extremely long
serum elimination half-life (up to 100 hours in patients with normal renal
function). The dose of teicoplanin in adults is 6 to 30 mg/kg per day, with
the higher dosages reserved for treatment of serious staphylococcal
infections. Once-daily dosing is possible for the treatment of most
infections because of the prolonged serum elimination half-life. As with
vancomycin, teicoplanin doses must be adjusted in patients with renal
insufficiency. For functionally anephric patients, administration once weekly
has been appropriate, but serum drug concentrations should be monitored to
determine that the therapeutic range has been maintained (e.g., trough
concentration of 15 to 20 Therapeutic Uses Teicoplanin has been used to treat a wide variety of infections, including osteomyelitis and endocarditis, caused by methicillin-resistant and methicillin-susceptible staphylococci, streptococci, and enterococci (Bibler et al., 1987; Glupczynski et al., 1986). Teicoplanin has been found to be comparable to vancomycin in efficacy, except for treatment failures from low doses used to treat serious infections, such as endocarditis (Calain et al., 1987). Teicoplanin is not as efficacious as antistaphylococcal penicillins for treating bacteremia and endocarditis caused by methicillin-susceptible S. aureus, with teicoplanin cure rates of 60% to 70% versus 85% to 90% for the penicillins. The efficacy of teicoplanin against S. aureus may be improved by the addition of an aminoglycoside (e.g., gentamicin 1 mg/kg every 8 hours in patients with normal renal function) to provide a synergistic effect. Strains of streptococci are uniformly susceptible to teicoplanin. This drug has been very effective in a once-daily regimen for patients with streptococcal osteomyelitis or endocarditis (Leport et al., 1989). Teicoplanin is among the most active drugs against enterococci. Limited experience indicates that it is effective, although only bacteriostatic, for serious enterococcal infections. It should be combined with gentamicin to achieve a bactericidal effect in the treatment of enterococcal endocarditis. Untoward Effects The main side effect reported for teicoplanin is skin rash, which is more common in higher dosages. Hypersensitivity reactions, drug fever, and neutropenia also have been reported. Ototoxicity has occurred rarely. |
Bacitracin
|
History and Source Bacitracin is an antibiotic produced by the Tracy-I strain of Bacillus subtilis, isolated in 1943 from the damaged tissue and street dirt debrided from a compound fracture in a young girl named Tracy; hence the name bacitracin. The history, properties, and uses of bacitracin have been reviewed by Meleney and Johnson (1949). Chemistry The bacitracins are a group of polypeptide antibiotics; multiple components have been demonstrated in the commercial products. The major constituent is bacitracin A. Its probable structural formula is as follows:
A unit of the antibiotic is equivalent to 26 Antibacterial Activity A variety of gram-positive cocci and bacilli, Neisseria, H. influenzae, and Treponema pallidum are sensitive to 0.1 U or less of bacitracin per milliliter. Actinomyces and Fusobacterium are inhibited by concentrations of 0.5 to 5 U/ml. Enterobacteriaceae, Pseudomonas, Candida spp., and Nocardia are resistant to the drug. Bacitracin inhibits bacterial cell-wall synthesis. Absorption, Fate, and Excretion While bacitracin has been employed parenterally in the past and injectable products are still available, current use is restricted primarily to topical application. Therapeutic Uses Bacitracin is available in ophthalmic and dermatologic ointments; the antibiotic also is available in the form of a powder for the preparation of topical solutions. The ointments are applied directly to the involved surface one or more times daily. A number of topical preparations of bacitracin to which neomycin or polymyxin or both have been added are available, and some contain the three antibiotics plus hydrocortisone. Topical bacitracin alone or in combination with other antimicrobial agents has no established value in the treatment of furunculosis, pyoderma, carbuncle, impetigo, and superficial and deep abscesses. For open infections such as infected eczema and infected dermal ulcers, the local application of the antibiotic may be of some help in eradicating sensitive bacteria. Bacitracin has an advantage over other antibiotics in that topical administration, even in an ointment, rarely produces hypersensitivity. Suppurative conjunctivitis and infected corneal ulcer respond well to the topical use of bacitracin when they are caused by susceptible bacteria. Bacitracin has been used with limited success for eradication of nasal carriage of staphylococci. Oral bacitracin has been used with some success for the treatment of antibiotic-associated diarrhea caused by C. difficile (Dudley et al., 1986). Untoward Effects Serious nephrotoxicity results from the parenteral use of this antibiotic. Hypersensitivity reactions result from topical application, but this is uncommon. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3768
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved