| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Diuretics
Overview
|
Diuretics increase the rate of urine flow and sodium excretion and are used to adjust the volume and/or composition of body fluids in a variety of clinical situations, including hypertension, heart failure, renal failure, nephrotic syndrome, and cirrhosis. The objective of this chapter is to provide the reader with unifying concepts as to how the kidney operates and how diuretics modify renal function. The chapter begins with a description of renal anatomy and physiology, as this information is prerequisite to a discussion of diuretic pharmacology. Categories of diuretics are introduced and then described with regard to chemistry, mechanism of action, site of action, effects on urinary composition, and effects on renal hemodynamics. Near the end of the chapter, diuretic pharmacology is integrated with a discussion of mechanisms of edema formation and the role of diuretics in clinical medicine. Therapeutic applications of diuretics are expanded upon in Chapters 33: Antihypertensive Agents and the Drug Therapy of Hypertension (hypertension) and 34: Pharmacological Treatment of Heart Failure (heart failure). |
Renal Anatomy and Physiology
|
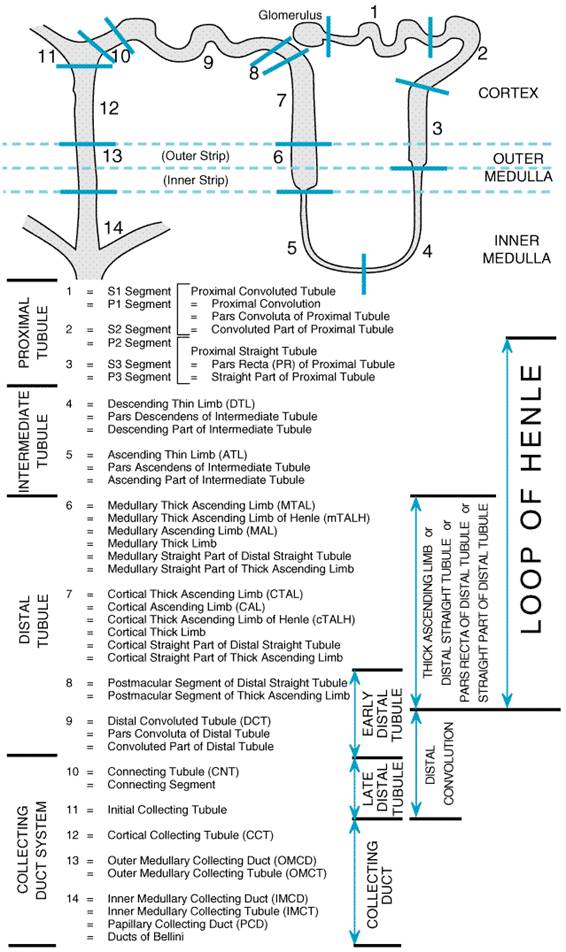
Renal Anatomy The main renal artery branches close to the renal hilum into segmental arteries, which, in turn, subdivide to form interlobar arteries that pierce the renal parenchyma. The interlobar arteries curve at the border of the renal medulla and cortex to form arc-like vessels known as arcuate arteries. Arcuate arteries give rise to perpendicular branches, called interlobular arteries, which enter the renal cortex and supply blood to the afferent arterioles. A single afferent arteriole penetrates the glomerulus of each nephron and branches extensively to form the glomerular capillary nexus. These branches coalesce to form the efferent arteriole. Efferent arterioles of superficial glomeruli ascend toward the kidney surface before splitting into peritubular capillaries that service the tubular elements of the renal cortex. Efferent arterioles of juxtamedullary glomeruli descend into the medulla and divide to form the descending vasa recta, which supply blood to the capillaries of the medulla. Blood returning from the medulla via the ascending vasa recta drains directly into the arcuate veins, and blood from the peritubular capillaries of the cortex enters the interlobular veins, which, in turn, connect with the arcuate veins. Arcuate veins drain into interlobar veins, which in turn drain into segmental veins, and blood leaves the kidney via the main renal vein. The basic urine-forming unit of the kidney is the nephron, which consists of a filtering apparatus, the glomerulus, connected to a long tubular portion that reabsorbs and conditions the glomerular ultrafiltrate. Each human kidney is composed of approximately 1 million nephrons. The nomenclature for segments of the tubular portion of the nephron has become increasingly complex as renal physiologists have subdivided the nephron into shorter and shorter named segments. These subdivisions initially were based on the axial location of the segments but increasingly have been based on the morphology of the epithelial cells lining the various nephron segments. Figure 291 illustrates the currently accepted subdivision of the nephron into 14 subsegments. Commonly encountered names that refer to these subsegments and to combinations of subsegments are included.
Glomerular Filtration In the glomerular capillaries, a portion of the plasma water is forced
through a filter that has three basic components: the fenestrated capillary
endothelial cells, a basement membrane lying just beneath the endothelial
cells, and the filtration slit diaphragms formed by the epithelial cells that
cover the basement membrane on its urinary space side. Solutes of small size
flow with filtered water (solvent drag) into the urinary (Bowman's) space,
whereas formed elements and macromolecules are retained by the filtration
barrier. For each nephron unit, the rate of filtration (single-nephron
glomerular filtration rate, SNGFR) is a function of the hydrostatic pressure
in the glomerular capillaries (PGC), the hydrostatic
pressure in Bowman's space (which can be equated with pressure in the
proximal tubule, PT), the mean colloid osmotic pressure in
the glomerular capillaries ( SNGFR = Kf[(PGC
( If PGCPT is defined as the
transcapillary hydraulic pressure difference ( SNGFR = Kf( This latter equation succinctly expresses the three major determinants
of SNGFR. However, each of these three determinants can be influenced by a
number of other variables. Kf is determined by the
physicochemical properties of the filtering membrane and by the surface area
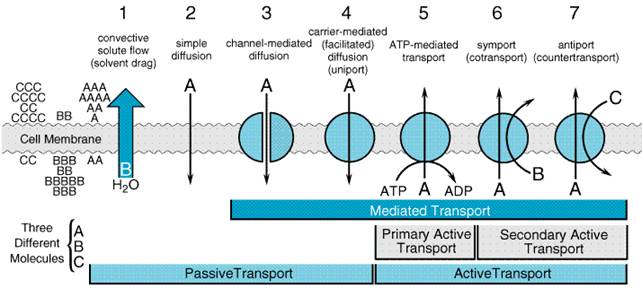
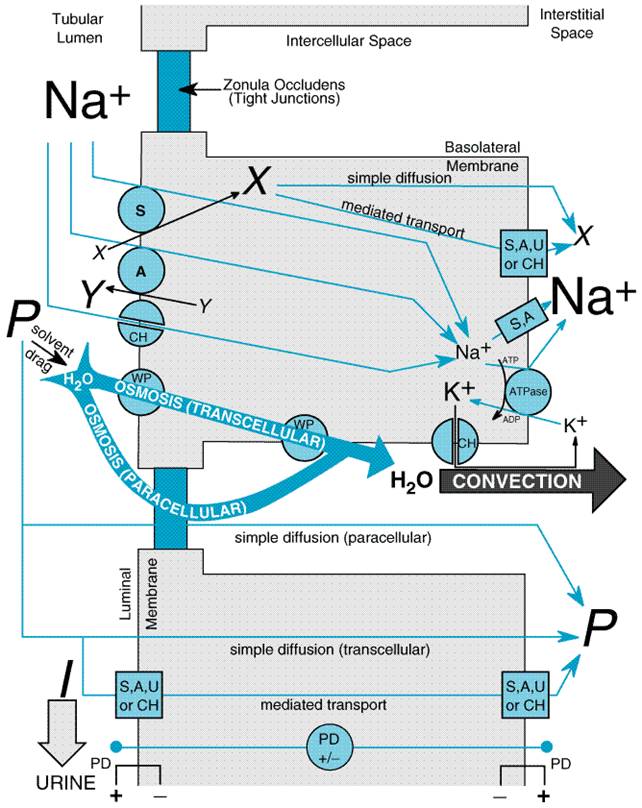
available for filtration. Overview of Nephron Function Approximately 120 ml of ultrafiltrate is formed each minute, yet only 1 ml/min of urine is produced. Therefore, greater than 99% of the glomerular ultrafiltrate is reabsorbed at a staggering energy cost. The kidneys consume 7% of total-body oxygen intake despite the fact that the kidneys make up only 0.5% of body weight. The kidney is designed to filter large quantities of plasma, reabsorb those substances that the body must conserve, and leave behind and/or secrete substances that must be eliminated. The proximal tubule is contiguous with Bowman's capsule and takes a tortuous path until finally forming a straight portion that dives into the renal medulla. The proximal tubule has been subdivided into S1, S2, and S3 segments based on the morphology of the epithelial cells lining the tubule. Normally, approximately 65% of filtered Na+ is reabsorbed in the proximal tubule, and since this part of the tubule is highly permeable to water, reabsorption is essentially isotonic. Between the outer and inner strips of the outer medulla, the tubule abruptly changes morphology to become the descending thin limb (DTL), which penetrates the inner medulla, makes a hairpin turn, and then forms the ascending thin limb (ATL). At the juncture between the inner and outer medulla, the tubule once again changes morphology and becomes the thick ascending limb, which is made up of three segments: a medullary portion (MTAL), a cortical portion (CTAL), and a postmacular segment. Together, the proximal straight tubule, DTL, ATL, MTAL, CTAL, and postmacular segment are known as the loop of Henle. The DTL is highly permeable to water, yet its permeability to NaCl and urea is low. In contrast, the ATL is permeable to NaCl and urea but is impermeable to water. The thick ascending limb actively reabsorbs NaCl but is impermeable to water and urea. Approximately 25% of filtered Na+ is reabsorbed in the loop of Henle, mostly in the thick ascending limb, which has a large reabsorptive capacity. The thick ascending limb passes between the afferent and efferent arterioles and makes contact with the afferent arteriole via a cluster of specialized columnar epithelial cells known as the macula densa. The macula densa is strategically located to sense concentrations of NaCl leaving the loop of Henle. If the concentration of NaCl is too high, the macula densa sends a chemical signal (perhaps adenosine) to the afferent arteriole of the same nephron, causing it to constrict. This in turn causes a reduction in PGC and QA and decreases SNGFR. This homeostatic mechanism, known as tubuloglomerular feedback (TGF), serves to protect the organism from salt and volume wasting. Besides causing a TGF response, the macula densa also regulates renin release from the adjacent juxtaglomerular cells in the wall of the afferent arteriole. Approximately 0.2 mm past the macula densa, the tubule changes morphology once again to become the distal convoluted tubule (DCT). The postmacular segment of the thick ascending limb and the distal convoluted tubule often are referred to as the early distal tubule. Like the thick ascending limb, the DCT actively transports NaCl and is impermeable to water. Since these characteristics impart the ability to produce a dilute urine, the thick ascending limb and the DCT are collectively called the diluting segment of the nephron, and the tubular fluid in the DCT is hypotonic regardless of hydration status. However, unlike the thick ascending limb, the DCT does not contribute to the countercurrent-induced hypertonicity of the medullary interstitium (see below). The collecting duct system (connecting tubule + initial collecting tubule + cortical collecting duct + outer and inner medullary collecting duct) is an area of fine control of ultrafiltrate composition and volume. It is here that final adjustments in electrolyte composition are made, a process modulated by the adrenal steroid, aldosterone. In addition, permeability of this part of the nephron to water is modulated by antidiuretic hormone (ADH; seeChapter 30: Vasopressin and Other Agents Affecting the Renal Conservation of Water). The more distal portions of the collecting duct pass through the renal medulla, where the interstitial fluid is markedly hypertonic. In the absence of ADH, the collecting duct system is impermeable to water, and a dilute urine is excreted. However, in the presence of ADH, the collecting duct system is permeable to water, so that water is reabsorbed. The movement of water out of the tubule is driven by the steep concentration gradient that exists between the tubular fluid and the medullary interstitium. The hypertonicity of the medullary interstitium plays a vital role in the ability of mammals and birds to concentrate urine and is therefore a key adaptation necessary for living in a terrestrial environment. This is accomplished via a combination of the unique topography of the loop of Henle and the specialized permeability features of the loop's subsegments. Although the precise mechanism giving rise to the medullary hypertonicity has remained elusive, the passive countercurrent multiplier hypothesis of Kokko and Rector (1972) is an intuitively attractive model that is qualitatively accurate (seeSands and Kokko, 1996). According to this hypothesis, the process begins with active transport in the thick ascending limb, which concentrates NaCl in the interstitium of the outer medulla. Since this segment of the nephron is impermeable to water, active transport in the ascending limb dilutes the tubular fluid. As the dilute fluid passes into the collecting duct system, water is extracted if and only if ADH is present. Since the cortical and outer medullary collecting ducts have a low permeability to urea, urea is concentrated in the tubular fluid. The inner medullary collecting duct, however, is permeable to urea, so that urea diffuses into the inner medulla where it is trapped by countercurrent exchange in the vasa recta. Since the DTL is impermeable to salt and urea, the high urea concentration in the inner medulla extracts water from the DTL and concentrates NaCl in the tubular fluid of the DTL. As the tubular fluid enters the ATL, NaCl diffuses out of the salt-permeable ATL, thus contributing to the hypertonicity of the medullary interstitium. General Mechanism of Renal Epithelial Transport Figure 292 illustrates seven mechanisms by which solute crosses renal epithelial cell membranes. If bulk water flow occurs across a membrane, solute molecules will be transferred by convection across the membrane, a process known as solvent drag. Solutes with sufficient lipid solubility may also dissolve in the membrane and diffuse across the membrane down their electrochemical gradients (simple diffusion). Many solutes, however, have limited lipid solubility, and transport must rely on integral proteins embedded in the cell membrane. In some cases, the integral protein merely provides a conductive pathway (pore) through which the solute may diffuse passively (channel-mediated diffusion). In other cases, the solute may bind to the integral protein and, due to a conformational change in the protein, be transferred across the cell membrane down an electrochemical gradient (carrier-mediated or facilitated diffusion, also called uniport). However, this process will not result in net movement of solute against an electrochemical gradient. If solute must be moved 'uphill' against an electrochemical gradient, then either primary active transport or secondary active transport is required. With primary active transport, ATP hydrolysis is coupled directly to conformational changes in the integral protein, thus providing the necessary free energy (ATP-mediated transport). Often, ATP-mediated transport is used to create an electrochemical gradient for a given solute, and the free energy of that solute gradient is then released to drive the 'uphill' transport of other solutes. This process requires symport (cotransport of solute species in the same direction) or antiport (countertransport of solute species in opposite directions) and is known as secondary active transport.
The kinds of transport achieved in a particular nephron segment depend mainly on which transporters are present and whether they are embedded in the luminal or basolateral membrane. A general model of renal tubular transport is shown in Figure 293 and can be summarized as follows:
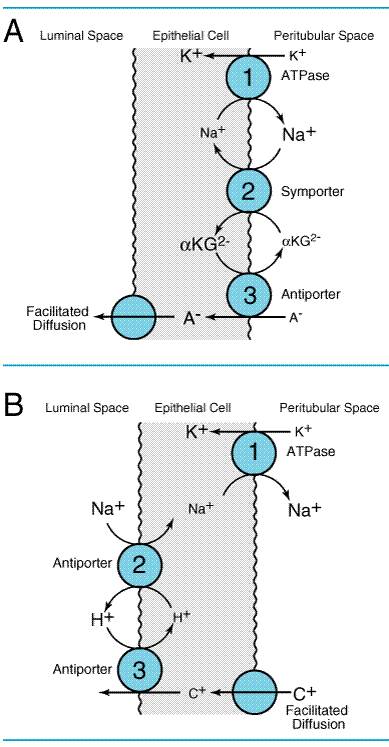
Mechanism of Organic Acid and Organic Base Secretion The kidney is a major organ involved in the elimination of organic
chemicals from the body. Organic molecules may enter the renal tubules by
glomerular filtration of molecules not bound to plasma proteins or may be
actively secreted directly into the tubules. The proximal tubule has a highly
efficient transport system for organic acids and an equally efficient but
separate transport system for organic bases. Current models for these
secretory systems are illustrated in Figure 294. Both systems are powered by
the sodium pump in the basolateral membrane, involve secondary and tertiary
active transport, and utilize a poorly characterized facilitated-diffusion
step. The antiporter that exchanges
Renal Handling of Specific Anions and Cations Reabsorption of Cl generally follows reabsorption of Na+. In segments of the tubule with low-resistance tight junctions (i.e., 'leaky' epithelium), such as the proximal tubule and thick ascending limb, Cl movement can occur paracellularly. With regard to transcellular Cl flux, Cl crosses the luminal membrane via antiport with formate and oxalate (proximal tubule), symport with Na+/K+ (thick ascending limb), symport with Na+ (DCT), and antiport with HCO3 (collecting duct system). Cl crosses the basolateral membrane via symport with K+ (proximal tubule and thick ascending limb), antiport with Na+/HCO3 (proximal tubule), and Cl channels (thick ascending limb, DCT, collecting duct system). Eighty to ninety percent of filtered K+ is reabsorbed in the proximal tubule (diffusion and solvent drag) and thick ascending limb (diffusion), largely via the paracellular pathway. In contrast, the DCT and collecting duct system secrete variable amounts of K+via a conductive (channel-mediated) pathway. Modulation of the rate of K+ secretion in the collecting duct system, particularly by aldosterone, allows urinary excretion of K+ to be matched with dietary intake. The transepithelial potential difference (VT), lumen-positive in the thick ascending limb and lumen-negative in the collecting duct system, provides an important driving force for K+ reabsorption and secretion, respectively. Most of the filtered Ca2+ (approximately 70%) is reabsorbed by the proximal tubule by passive diffusion, probably via a paracellular route. Another 25% of filtered Ca2+ is reabsorbed by the thick ascending limb, mostly via a paracellular route driven by the lumen-positive VT, although a component of active Ca2+ reabsorption also may exist. The remaining Ca2+ is reabsorbed in the distal convoluted tubule and the connecting tubule via a transcellular pathway that is modulated by parathyroid hormone (PTH; seeChapter 62: Agents Affecting Calcification and Bone Turnover: Calcium, Phosphate, Parathyroid Hormone, Vitamin D, Calcitonin, and Other Compounds). PTH appears to increase Ca2+ channels in the luminal membrane, thereby facilitating the passive movement of Ca2+ into the epithelial cell. Ca2+ is extruded across the basolateral membrane by a Ca2+ATPase and via Na+Ca2+ antiport. Inorganic phosphate (Pi) is largely reabsorbed (80% of filtered load) by the proximal tubule. A Na+Pi symporter uses the free energy of the Na+ electrochemical gradient to effect secondary active transport of Pi into the cell. The Na+Pi symporter is inhibited by PTH. Pi exits the basolateral membrane down its electrochemical gradient by a poorly understood transport system. Only 20% to 25% of Mg2+ is reabsorbed in the proximal tubule, and only 5% is reabsorbed by the DCT and collecting duct system. The bulk of Mg2+ is reabsorbed in the thick ascending limb via a paracellular pathway driven by the lumen-positive VT. However, transcellular movement of Mg2+ also may occur with basolateral exit via Na+Mg2+ antiport or via a Mg2+ATPase. The renal tubules play an extremely important role in the reabsorption of HCO3 and secretion of protons (tubular acidification) and thus participate critically in the maintenance of acidbase balance. A description of these processes is presented in the section on carbonic anhydrase inhibitors. |
Principles of Diuretic Action
|
By definition, diuretics are drugs that increase the rate of urine flow; however, clinically useful diuretics also increase the rate of excretion of Na+ (natriuresis) and of an accompanying anion, usually Cl. NaCl in the body is the major determinant of extracellular fluid volume, and most clinical applications of diuretics are directed toward reducing extracellular fluid volume by decreasing total-body NaCl content. A sustained imbalance between dietary Na+ intake and Na+ loss is incompatible with life. A sustained positive Na+ balance would result in volume overload with pulmonary edema, and a sustained negative Na+ balance would result in volume depletion and cardiovascular collapse. Although continued administration of a diuretic causes a sustained net deficit in total-body Na+, the time course of natriuresis is finite as renal compensatory mechanisms bring Na+ excretion in line with Na+ intake, a phenomenon known as 'diuretic braking.' These compensatory, or braking, mechanisms include activation of the sympathetic nervous system, activation of the reninangiotensinaldosterone axis, decreased arterial blood pressure (which reduces pressure-natriuresis), hypertrophy of renal epithelial cells, increased expression of renal epithelial transporters, and perhaps alterations in natriuretic hormones such as atrial natriuretic peptide. Historically, the classification of diuretics was based on a mosaic of ideas such as site of action (loop diuretics), efficacy (high-ceiling diuretics), chemical structure (thiazide diuretics), similarity of action with other diuretics (thiazide-like diuretics), effects on potassium excretion (potassium-sparing diuretics), etc. However, since the mechanism of action of each of the major classes of diuretics is now reasonably well understood, a classification scheme based on mechanism of action is now possible and is used in this chapter. Diuretics not only alter the excretion of Na+, but also may modify renal handling of other cations (e.g., K+, H+, Ca2+, and Mg2+), anions (e.g., Cl, HCO3, and H2PO4), and uric acid. In addition, diuretics may indirectly alter renal hemodynamics. Table 291 gives a comparison of the general effects of the major classes of diuretics. |
Inhibitors of Carbonic Anhydrase
|
Acetazolamide DIAMOX) is the prototype of a class of agents that have limited usefulness as diuretics but have played a major role in the development of fundamental concepts of renal physiology and pharmacology. Chemistry When sulfanilamide was introduced as a chemotherapeutic agent,
metabolic acidosis was recognized as a side effect. This observation led to in
vitro and in vivo studies demonstrating that sulfanilamide is an
inhibitor of carbonic anhydrase. Subsequently, an enormous number of
sulfonamides were synthesized and tested for the ability to inhibit carbonic
anhydrase; of these compounds, acetazolamide has been most extensively studied.
Table 292 lists the chemical structures of the three carbonic anhydrase
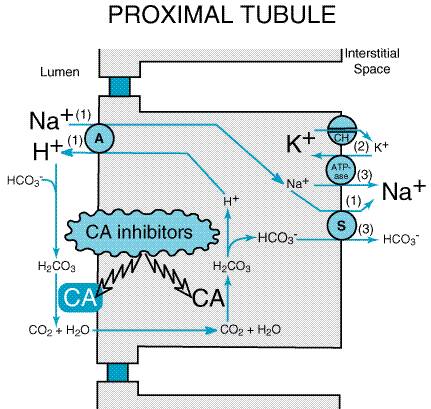
inhibitors currently available in the Mechanism and Site of Action Proximal tubular epithelial cells are richly endowed with the zinc metalloenzyme carbonic anhydrase, which is found in the luminal and basolateral membranes (type IV carbonic anhydrase, an enzyme tethered to the membrane by a glycosylphosphatidylinositol linkage) as well as in the cytoplasm (type II carbonic anhydrase). Davenport and Wilhelmi (1941) were the first to discover this enzyme in the mammalian kidney, and subsequent studies revealed the key role played by carbonic anhydrase in NaHCO3 reabsorption and acid secretion (seeMaren, 1967 and 1980). In the proximal tubule, the free energy in the Na+ gradient
established by the basolateral Na+ pump is used by a Na+H+
antiporter (also referred to as a Na+H+ exchanger or
NHE) in the luminal membrane to transport H+ into the tubular
lumen in exchange for Na+ (Figure 295). In the lumen, H+
reacts with filtered HCO3 to form H2CO3,
which rapidly decomposes to CO2 and water in the presence of
carbonic anhydrase in the brush border. Normally the reaction between CO2
and water occurs slowly, but carbonic anhydrase reversibly accelerates this
reaction several thousandfold. CO2 is lipophilic and rapidly
diffuses across the luminal membrane into the epithelial cell where it reacts
with water to form H2CO3, a reaction catalyzed by
cytoplasmic carbonic anhydrase. (The actual reaction catalyzed by carbonic
anhydrase is
Carbonic anhydrase inhibitors potently inhibit (IC50 for acetazolamide is 10 nM) both the membrane-bound and cytoplasmic forms of carbonic anhydrase, resulting in nearly complete abolition of NaHCO3 reabsorption in the proximal tubule (Cogan et al., 1979). Studies with a high-molecular-weight carbonic anhydrase inhibitor that only inhibits luminal enzyme because of limited cellular permeability indicate that inhibition of both the membrane-bound and cytoplasmic pools of carbonic anhydrase contributes to the diuretic activity of carbonic anhydrase inhibitors (Maren et al., 1997). Because of the large excess of carbonic anhydrase in proximal tubules, a high percentage of enzyme activity must be inhibited before an effect on electrolyte excretion is observed. Although the proximal tubule is the major site of action of carbonic anhydrase inhibitors, carbonic anhydrase also is involved in secretion of titratable acid in the collecting duct system (a process that involves a proton pump); therefore the collecting duct system is a secondary site of action for this class of drugs. Effects on Urinary Excretion Inhibition of carbonic anhydrase is associated with a rapid rise in urinary HCO3 excretion to approximately 35% of filtered load. This, along with inhibition of titratable acid and ammonia secretion in the collecting duct system, results in an increase in urinary pH to approximately 8 and development of a metabolic acidosis. However, even with a high degree of inhibition of carbonic anhydrase, 65% of HCO3 is rescued from excretion by poorly understood mechanisms that may involve carbonic anhydraseindependent HCO3 reabsorption at downstream sites. Inhibition of the transport mechanism described in the preceding section results in increased delivery of Na+ and Cl to the loop of Henle, which has a large reabsorptive capacity and captures most of the Cl and a portion of the Na+. Thus, only a small increase in Cl excretion occurs, HCO3 being the major anion excreted along with the cations Na+ and K+. The fractional excretion of Na+ may be as much as 5%, and the fractional excretion of K+ can be as much as 70%. The increased excretion of K+ is secondary to increased delivery of Na+ to the distal nephron. The mechanism by which increased distal delivery of Na+ enhances K+ excretion is described in the section on inhibitors of sodium channels. Carbonic anhydrase inhibitors also increase phosphate excretion (mechanism unknown), but have little or no effect on the excretion of Ca2+ or Mg2+. The effects of carbonic anhydrase inhibitors on renal excretion are self-limiting, probably because, as metabolic acidosis develops, the filtered load of HCO3 decreases to the point that the uncatalyzed reaction between CO2 and water is sufficient to achieve HCO3 reabsorption. Effects on Renal Hemodynamics By inhibiting proximal reabsorption, carbonic anhydrase inhibitors increase delivery of solutes to the macula densa. This triggers tubuloglomerular feedback (TGF), which increases afferent arteriolar resistance and reduces renal blood flow (RBF) and glomerular filtration rate (GFR) (Persson and Wright, 1982). Other Actions Carbonic anhydrase is present in a number of extrarenal tissues including the eye, gastric mucosa, pancreas, central nervous system (CNS), and red blood cells (RBCs). Carbonic anhydrase in the ciliary processes of the eye mediates the formation of large amounts of HCO3 in aqueous humor. For this reason, inhibition of carbonic anhydrase decreases the rate of formation of aqueous humor and consequently reduces intraocular pressure. Acetazolamide frequently causes paresthesias and somnolence, suggesting an action of carbonic anhydrase inhibitors in the CNS. The efficacy of acetazolamide in epilepsy is in part due to the production of metabolic acidosis; however, direct actions of acetazolamide in the CNS also contribute to its anticonvulsant action. Due to interference with carbonic anhydrase activity in RBCs, carbonic anhydrase inhibitors increase CO2 levels in peripheral tissues and decrease CO2 levels in expired gas. Large doses of carbonic anhydrase inhibitors reduce gastric acid secretion, but this has no therapeutic applications. Absorption and Elimination The oral bioavailability, plasma half-life, and route of elimination of the three currently available carbonic anhydrase inhibitors are listed in Table 292. Carbonic anhydrase inhibitors are avidly bound by carbonic anhydrase and, accordingly, tissues rich in this enzyme will have higher concentrations of carbonic anhydrase inhibitors following systemic administration. Toxicity, Adverse Effects, Contraindications, Drug Interactions Serious toxic reactions to carbonic anhydrase inhibitors are infrequent; however, these drugs are sulfonamide derivatives and, like other sulfonamides, may cause bone-marrow depression, skin toxicity, and sulfonamide-like renal lesions and may cause allergic reactions in patients hypersensitive to sulfonamides. With large doses, many patients exhibit drowsiness and paresthesias. Most adverse effects, contraindications, and drug interactions are secondary to urinary alkalinization or metabolic acidosis, including: (1) diversion of ammonia of renal origin from urine into the systemic circulation, a process that may induce hepatic encephalopathy (the drugs are contraindicated in patients with hepatic cirrhosis); (2) calculus formation and ureteral colic due to precipitation of calcium phosphate salts in an alkaline urine; (3) worsening of metabolic or respiratory acidosis (the drugs are contraindicated in patients with hyperchloremic acidosis or severe chronic obstructive pulmonary disease); (4) interference with the urinary tract antiseptic methenamine; and (5) reduction of the urinary excretion rate of weak organic bases. Therapeutic Uses Although acetazolamide is used for treatment of edema, the efficacy of carbonic anhydrase inhibitors as single agents is low, and carbonic anhydrase inhibitors are not widely employed in this regard. However, studies by Knauf and Mutschler (1997) indicate that the combination of acetazolamide with diuretics that block Na+ reabsorption at more distal sites in the nephron causes a marked natriuretic response in patients with low basal fractional excretion of Na+ (<0.2%) who are resistant to diuretic monotherapy. Even so, the long-term usefulness of carbonic anhydrase inhibitors often is compromised by development of metabolic acidosis. The major indication for carbonic anhydrase inhibitors is open-angle glaucoma. Carbonic anhydrase inhibitors also may be employed for secondary glaucoma and preoperatively in acute angle-closure glaucoma to lower ocular pressure before surgery (seeChapter 66: Ocular Pharmacology). Acetazolamide also is used for the treatment of epilepsy (seeChapter 21: Drugs Effective in the Therapy of the Epilepsies). The rapid development of tolerance, however, may limit the usefulness of carbonic anhydrase inhibitors for epilepsy. Acetazolamide may provide symptomatic relief in patients with acute mountain sickness; however, it is more appropriate to give acetazolamide as a prophylactic measure (Coote, 1991). Acetazolamide also is useful in patients with familial periodic paralysis (Links et al., 1988). The mechanism for the beneficial effects of acetazolamide in mountain sickness and familial periodic paralysis is not clear, but it may be related to the induction of a metabolic acidosis. Finally, carbonic anhydrase inhibitors can be useful for correcting a metabolic alkalosis, especially an alkalosis caused by diuretic-induced increases in H+ excretion. |
Osmotic Diuretics
|
Osmotic diuretics are agents that are freely filtered at the glomerulus, undergo limited reabsorption by the renal tubule, and are relatively inert pharmacologically. Osmotic diuretics are administered in large enough doses to increase significantly the osmolality of plasma and tubular fluid. Table 293 gives the molecular structures of the four currently available osmotic diureticsglycerin, isosorbide, mannitol, and urea. Mechanism and Site of Action For many years it was thought that osmotic diuretics act primarily in the proximal tubule (Wesson and Anslow, 1948). By acting as nonreabsorbable solutes, it was reasoned that osmotic diuretics limit the osmosis of water into the interstitial space and thereby reduce luminal Na+ concentration to the point that net Na+ reabsorption ceases. Indeed, early micropuncture studies supported this concept (Windhager et al., 1959). However, subsequent studies suggest that this mechanism, while operative, may be of only secondary importance. For instance, mannitol only slightly increases the delivery of Na+ and moderately increases the delivery of water out of the proximal tubule (Seely and Dirks, 1969), and urea does not alter proximal tubular reabsorption in rats at the time of a large osmotic diuresis (Kauker et al., 1970). Mannitol, on the other hand, markedly increases the delivery of Na+ and water out of the loop of Henle (Seely and Dirks, 1969), suggesting that the major site of action is the loop of Henle. By extracting water from intracellular compartments, osmotic diuretics expand the extracellular fluid volume, decrease blood viscosity, and inhibit renin release. These effects increase RBF, and the increase in renal medullary blood flow removes NaCl and urea from the renal medulla, thus reducing medullary tonicity. Also, under some circumstances, prostaglandins may contribute to the renal vasodilation and medullary washout induced by osmotic diuretics (Johnston et al., 1981). A reduction in medullary tonicity causes a decrease in the extraction of water from the DTL, which in turn limits the concentration of NaCl in the tubular fluid entering the ATL. This latter effect diminishes the passive reabsorption of NaCl in the ATL. In addition, the marked ability of osmotic diuretics to inhibit reabsorption of Mg2+, a cation that is mainly reabsorbed in the thick ascending limb, suggests that osmotic diuretics also interfere with transport processes in the thick ascending limb. The mechanism of this effect is unknown. In summary, osmotic diuretics act both in the proximal tubule and the loop of Henle, with the latter being the primary site of action. Also, osmotic diuretics probably act by an osmotic effect in the tubules and by reducing medullary tonicity. Effects on Urinary Excretion Osmotic diuretics increase the urinary excretion of nearly all electrolytes, including Na+, K+, Ca2+, Mg2+, Cl, HCO3, and phosphate. Effects on Renal Hemodynamics As indicated in the preceding section, osmotic diuretics increase RBF
by a variety of mechanisms. Osmotic diuretics dilate the afferent arteriole,
which increases PGC, and dilute the plasma, which decreases
Absorption and Elimination The oral bioavailability, plasma half-life, and route of elimination of the four currently available osmotic diuretics are listed in Table 293. Glycerin and isosorbide can be given orally, whereas mannitol and urea must be administered intravenously. Toxicity, Adverse Effects, Contraindications, Drug Interactions Osmotic diuretics are distributed in the extracellular fluid and contribute to the extracellular osmolality. Thus, water is extracted from intracellular compartments, and the extracellular fluid volume becomes expanded. In patients with heart failure or pulmonary congestion, this may cause frank pulmonary edema. Extraction of water also causes hyponatremia, which may explain common adverse effects, including headache, nausea, and vomiting. On the other hand, loss of water in excess of electrolytes can cause hypernatremia and dehydration. In general, osmotic diuretics are contraindicated in patients who are anuric due to severe renal disease or who are unresponsive to test doses of the drugs. Urea may cause thrombosis or pain if extravasation occurs, and it should not be administered to patients with impaired liver function because of the risk of elevation of blood ammonia levels. Both mannitol and urea are contraindicated in patients with active cranial bleeding. Glycerin is metabolized and can cause hyperglycemia. Therapeutic Uses A rapid decrease in GFR, i.e., acute renal failure (ARF), is a
serious medical condition that occurs in 5% of hospitalized patients and is
associated with a significant mortality rate. ARF can be caused by diverse
conditions both extrinsic (prerenal and postrenal failure) and intrinsic to the
kidney. Acute tubular necrosis (ATN), i.e., damage to tubular
epithelial cells, accounts for the majority of cases of intrinsic ARF. In
animal models, mannitol is effective in attenuating the reduction in GFR associated
with ATN when administered before the ischemic insult or offending
nephrotoxin. The renal protection afforded by mannitol may be due to removal
of obstructing tubular casts, dilution of nephrotoxic substances in the
tubular fluid, and/or reduction of swelling of tubular elements via
osmotic extraction of water. Although prophylactic mannitol is effective in
animal models of ATN, the clinical efficacy of mannitol is less well
established. Most published clinical studies have been uncontrolled, and
controlled studies have not shown a benefit over hydration per se (seeKellum,
1998). In patients with mild-to-moderate renal insufficiency, hydration with
0.45% sodium chloride is as good as or better than either mannitol or furosemide
in protection against decreases in GFR induced by radiocontrast agents
(Soloman et al., 1994). Studies of prophylactic mannitol indicate
effectiveness in jaundiced patients undergoing surgery ( Another use for mannitol and urea is in the treatment of dialysis disequilibrium syndrome. Too rapid a removal of solutes from the extracellular fluid by hemodialysis or peritoneal dialysis results in a reduction in the osmolality of the extracellular fluid. Consequently, water moves from the extracellular compartment into the intracellular compartment, causing hypotension and CNS symptoms (headache, nausea, muscle cramps, restlessness, CNS depression, and convulsions). Osmotic diuretics increase the osmolality of the extracellular fluid compartment and thereby shift water back into the extracellular compartment. By increasing the osmotic pressure of the plasma, osmotic diuretics extract water from the eye and brain. All four osmotic diuretics are used to control intraocular pressure during acute attacks of glaucoma and for short-term reductions in intraocular pressure, both preoperatively and postoperatively, in patients who require ocular surgery. Also, mannitol and urea are used to reduce cerebral edema and brain mass before and after neurosurgery. |
Inhibitors
of Na +K+2 Cl Symport (
|
Inhibitors of Na+K+2Cl symport are a group of diuretics that have in common an ability to block the Na+K+2Cl symporter in the thick ascending limb of the loop of Henle; hence these diuretics also are referred to as loop diuretics. Although the proximal tubule reabsorbs approximately 65% of the filtered Na+, diuretics acting only in the proximal tubule have limited efficacy because the thick ascending limb has a great reabsorptive capacity and reabsorbs most of the rejectate from the proximal tubule. Diuretics acting predominantly at sites past the thick ascending limb also have limited efficacy, because only a small percentage of the filtered Na+ load reaches these more distal sites. In contrast, inhibitors of Na+K+2Cl symport are highly efficacious, and for this reason they often are called high-ceiling diuretics. The efficacy of inhibitors of Na+K+2Cl symport in the thick ascending limb of the loop of Henle is due to a combination of two factors: (1) Approximately 25% of the filtered Na+ load normally is reabsorbed by the thick ascending limb; and (2) nephron segments past the thick ascending limb do not possess the reabsorptive capacity to rescue the flood of rejectate exiting the thick ascending limb. Chemistry Inhibitors of Na+K+2Cl symport are
a chemically diverse group of drugs (seeTable 294). Furosemide, bumetanide,
azosemide, piretanide, and tripamide all contain a sulfonamide moiety,
whereas ethacrynic acid is a phenoxyacetic acid derivative. Muzolimine has
neither of these structural features, and torsemide is a sulfonylurea. Only furosemide
(
LASIX ), bumetanide
(
BUMEX ), ethacrynic
acid (EDECRIN), and torsemide
(
DEMADEX ) are
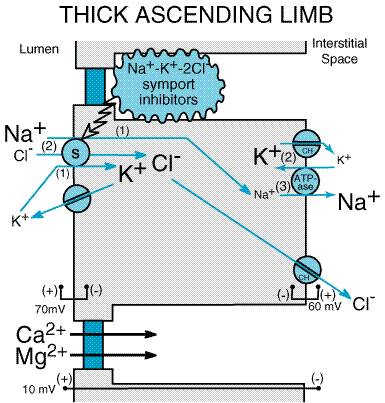
available currently in the Mechanism and Site of Action Inhibitors of Na+K+2Cl symport act primarily in the thick ascending limb. Micropuncture of the DCT demonstrates that loop diuretics increase the delivery of solutes out of the loop of Henle (Dirks and Seely, 1970). Also, in situ microperfusion of the loop of Henle (Morgan et al., 1970) and in vitro microperfusion of the CTAL (Burg et al., 1973) indicate inhibition of transport by low concentrations of furosemide in the perfusate. Some inhibitors of Na+K+2Cl symport may have additional effects in the proximal tubule; however, the significance of these effects is unclear. It was initially thought that Cl was transported by a primary active electrogenic transporter in the luminal membrane independent of Na+. Discovery of furosemide-sensitive Na+K+2Cl symport in other tissues caused Greger (1981) to investigate more carefully the Na+ dependence of Cl transport in the isolated perfused rabbit CTAL. By scrupulously removing Na+ from the luminal perfusate, Greger demonstrated the dependence of Cl transport on Na+. It is now well accepted that, in the thick ascending limb, flux of Na+, K+, and Cl from the lumen into the epithelial cell is mediated by a Na+K+2Cl symporter (seeFigure 296). This symporter captures the free energy in the Na+ electrochemical gradient established by the basolateral Na+ pump and provides for 'uphill' transport of K+ and Cl into the cell. K+ channels in the luminal membrane (called ROMK) provide a conductive pathway for the apical recycling of this cation (Ho et al., 1993; Kohda et al., 1998), and basolateral Cl channels (called CLCN) provide a basolateral exit mechanism for Cl. The luminal membranes of epithelial cells in the thick ascending limb have conductive pathways (channels) only for K+; therefore the apical membrane voltage is determined by the equilibrium potential for K+ (EK). In contrast, the basolateral membrane has channels for both K+ and Cl, so that the basolateral membrane voltage is less than EK; i.e., conductance for Cl depolarizes the basolateral membrane. Depolarization of the basolateral membrane results in a transepithelial potential difference of approximately 10 mV, with the lumen positive with respect to the interstitial space. This lumen-positive potential difference repels cations (Na+, Ca2+, and Mg2+) and thereby provides an important driving force for the paracellular flux of these cations into the interstitial space.
As the name implies, inhibitors of Na+K+2Cl symport bind to the Na+K+2Cl symporter in the thick ascending limb (Koenig et al., 1983) and block its function, bringing salt transport in this segment of the nephron to a virtual standstill (Burg et al., 1973). The molecular mechanism by which this class of drugs blocks the Na+K+2Cl symporter is unknown, but evidence suggests that these drugs attach to the Clbinding site (Hannafin et al., 1983) located in the symporter's transmembrane domain (Isenring and Forbush, 1997). Inhibitors of Na+K+2Cl symport also inhibit Ca2+ and Mg2+ reabsorption in the thick ascending limb by abolishing the transepithelial potential difference that is the dominant driving force for reabsorption of these cations. Na+K+2Cl symporters are an important family of transport molecules found in many secretory and absorbing epithelia. The rectal gland of the dogfish shark is a particularly rich source of the protein, and a cDNA encoding a Na+K+2Cl symporter was isolated from a cDNA library obtained from the dogfish shark rectal gland by screening with antibodies to the shark symporter (Xu et al., 1994). Molecular cloning revealed a deduced amino acid sequence of 1191 residues containing 12 putative membrane-spanning domains flanked by long N and C termini in the cytoplasm. Expression of this protein resulted in Na+K+2Cl symport that was sensitive to bumetanide. The shark rectal gland Na+K+2Cl symporter cDNA subsequently was used to screen a human colonic cDNA library, and this provided Na+K+2Cl symporter cDNA probes from this tissue. These latter probes were used to screen rabbit renal cortical and renal medullary libraries, which allowed cloning of the rabbit renal Na+K+2Cl symporter (Payne and Forbush, 1994). This symporter is 1099 amino acids in length, is 61% identical to the dogfish shark secretory Na+K+2Cl symporter, has 12 predicted transmembrane helices, and contains large N- and C-terminal cytoplasmic regions. Subsequent studies demonstrated that Na+K+2Cl symporters are of two varieties (seeKaplan et al., 1996). The 'absorptive' symporter (called ENCC2, NKCC2, or BSC1) is expressed only in the kidney, is localized to the apical membrane of the thick ascending limb, and is regulated by cyclic AMP (Obermller et al., 1996; Kaplan et al., 1996; Nielsen et al., 1998; Plata et al., 1999). At least six different isoforms of the absorptive symporter are generated by alternative mRNA splicing (Mount et al., 1999). The 'secretory' symporter (called ENCC3, NKCC1, or BSC2) is a 'housekeeping' protein that is widely expressed and, in epithelial cells, is localized to the basolateral membrane. A model of Na+K+2Cl symport has been proposed based on ordered binding of ions to the symporter (Lytle et al., 1998). Mutations in the genes coding for the absorptive Na+K+2Cl symporter, the apical K+ channel, or the basolateral Cl channel give rise to Bartter's syndrome (inherited hypokalemic alkalosis with salt wasting and hypotension) (seeSimon and Lifton, 1998). Effects on Urinary Excretion Due to blockade of the Na+K+2Cl symporter, loop diuretics cause a profound increase in the urinary excretion of Na+ and Cl (i.e., up to 25% of the filtered load of Na+). Abolition of the transepithelial potential difference also results in marked increases in the excretion of Ca2+ and Mg2+. Some (e.g., furosemide), but not all (e.g., bumetanide and piretanide), sulfonamide-based loop diuretics have weak carbonic anhydraseinhibiting activity. Those drugs with carbonic anhydraseinhibiting activity increase the urinary excretion of HCO3 and phosphate. The mechanism by which inhibition of carbonic anhydrase increases phosphate excretion is not known. All inhibitors of Na+K+2Cl symport increase the urinary excretion of K+ and titratable acid. This effect is due in part to increased delivery of Na+ to the distal tubule. The mechanism by which increased distal delivery of Na+ enhances excretion of K+ and H+ is discussed in the section on inhibitors of Na+ channels. Acutely, loop diuretics increase the excretion of uric acid, whereas chronic administration of these drugs results in reduced excretion of uric acid. The chronic effects of loop diuretics on uric acid excretion may be due to enhanced transport in the proximal tubule secondary to volume depletion, leading to increased uric acid reabsorption, or to competition between the diuretic and uric acid for the organic acid secretory mechanism in the proximal tubule, leading to reduced uric acid secretion. By blocking active NaCl reabsorption in the thick ascending limb, inhibitors of Na+K+2Cl symport interfere with a critical step in the mechanism that produces a hypertonic medullary interstitium. Therefore, loop diuretics block the kidney's ability to concentrate urine during hydropenia. Also, since the thick ascending limb is part of the diluting segment, inhibitors of Na+K+2Cl symport markedly impair the kidney's ability to excrete a dilute urine during water diuresis. Effects on Renal Hemodynamics If volume depletion is prevented by replacing fluid losses, inhibitors
of Na+K+2Cl symport generally increase
total RBF and redistribute RBF to the midcortex (Stein et al., 1972).
However, the effects on RBF are variable. The mechanism of the increase in
RBF is not known, but prostaglandins have been implicated (Williamson et
al., 1974). In fact, nonsteroidal antiinflammatory drugs (NSAIDs)
attenuate the diuretic response to loop diuretics, most likely by preventing
prostaglandin-mediated increases in RBF (Brater, 1985). Other Actions Absorption and Elimination The oral bioavailability, plasma half-life, and route of elimination
of the four inhibitors of Na+K+2Cl
symport available in the United States are listed in Table 294. Because furosemide,
bumetanide, ethacrynic acid, and torsemide are extensively bound to plasma
proteins, delivery of these drugs to the tubules by filtration is limited.
However, they are efficiently secreted by the organic acid transport system
in the proximal tubule and thereby gain access to their binding sites on the
Na+K+2Cl symport in the luminal membrane
of the thick ascending limb. Probenecid shifts the plasma
concentrationresponse curve to furosemide to the right by competitively
inhibiting furosemide secretion by the organic acid transport system (Brater,
1983). The most recent loop diuretic to receive FDA approval is torsemide,
which has a longer half-life than the other loop diuretics available in the Toxicity, Adverse Effects, Contraindications, Drug Interactions Adverse effects unrelated to the diuretic efficacy are rare, and most adverse effects are due to abnormalities of fluid and electrolyte balance. Overzealous use of loop diuretics can cause serious depletion of total body Na+. This may be manifest as hyponatremia and/or extracellular fluid volume depletion associated with hypotension, reduced GFR, circulatory collapse, thromboembolic episodes, and, in patients with liver disease, hepatic encephalopathy. Increased delivery of Na+ to the distal tubule, particularly when combined with activation of the reninangiotensin system, leads to increased urinary excretion of K+ and H+, causing a hypochloremic alkalosis. If dietary K+ intake is not sufficient, hypokalemia may develop, and this may induce cardiac arrhythmias, particularly in patients taking cardiac glycosides. Increased Mg2+ and Ca2+ excretion may result in hypomagnesemia (a risk factor for cardiac arrhythmias) and hypocalcemia (rarely leading to tetany). Contraindications to the use of loop diuretics include severe Na+ and volume depletion, hypersensitivity to sulfonamides (for sulfonamide-based loop diuretics), and anuria unresponsive to a trial dose of loop diuretic. Drug interactions may occur when loop diuretics are coadministered with: (1) aminoglycosides (synergism of ototoxicity caused by both drugs); (2) anticoagulants (increased anticoagulant activity); (3) digitalis glycosides (increased digitalis-induced arrhythmias); (4) lithium (increased plasma levels of lithium); (5) propranolol (increased plasma levels of propranolol); (6) sulfonylureas (hyperglycemia); (7) cisplatin (increased risk of diuretic-induced ototoxicity); (8) NSAIDs (blunted diuretic response; salicylate toxity when given with high doses of salicylates); (9) probenecid (blunted diuretic response); (10) thiazide diuretics (synergism of diuretic activity of both drugs leading to profound diuresis); and (11) amphotericin B (increased potential for nephrotoxicity and toxicity and intensification of electrolyte imbalance). Therapeutic Uses A major use of loop diuretics is in the treatment of acute pulmonary
edema. A rapid increase in venous capacitance in conjunction with a brisk
natriuresis reduces left ventricular filling pressures and thereby rapidly
relieves pulmonary edema. |
Inhibitors of Na+ Cl Symport (Thiazide and Thiazide-Like Diuretics)
|
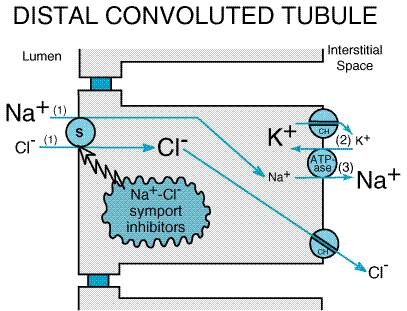
The benzothiadiazides were synthesized in an effort to enhance the potency of inhibitors of carbonic anhydrase. However, unlike carbonic anhydrase inhibitors, which primarily increase NaHCO3 excretion, benzothiadiazides were found predominantly to increase NaCl excretion (Beyer, 1958), an effect shown to be independent of carbonic anhydrase inhibition. Chlorothiazide was the first serious challenge to the mercurial diuretics, a now-obsolete class of organometallic compounds that dominated diuretic therapy for more than thirty years. Chemistry Inhibitors of Na+Cl symport are sulfonamides (seeTable 295) and many are analogs of 1,2,4-benzothiadiazine-1,1-dioxide. Because the original inhibitors of Na+Cl symport were benzothiadiazine derivatives, this class of diuretics became known as thiazide diuretics. Subsequently, drugs that are pharmacologically similar to thiazide diuretics but are not thiazides were developed and are called thiazide-like diuretics. The term thiazide diuretics often is used to refer to all members of the class of inhibitors of Na+Cl symport, and this usage is employed in the present chapter. Mechanism and Site of Action Some studies using split-droplet and stationary-microperfusion techniques have described reductions in proximal tubule reabsorption by thiazide diuretics; however, free-flow micropuncture studies have not consistently demonstrated increased solute delivery out of the proximal tubule following administration of thiazides. In contrast, micropuncture (Kunau et al., 1975) and in situ microperfusion studies (Costanzo and Windhager, 1978) clearly indicate that thiazide diuretics inhibit NaCl transport in the DCT. Furthermore, the renal cortex has a high-affinity receptor for thiazide diuretics (Beaumont et al., 1988), and binding of thiazides localizes to the DCT (Beaumont et al., 1989). It is now well accepted that the primary site of action of thiazide diuretics is the DCT, whereas the proximal tubule may represent a secondary site of action. Figure 297 illustrates the current model of electrolyte transport in the DCT. As with other nephron segments, transport is powered by a Na+ pump in the basolateral membrane. The free energy in the electrochemical gradient for Na+ is harnessed by a Na+Cl symporter in the luminal membrane, which moves Cl into the epithelial cell against its electrochemical gradient. Cl then passively exits the basolateral membrane via a Cl channel. Thiazide diuretics inhibit the Na+Cl symporter, perhaps by competing for the Cl binding site (Beaumont et al., 1988).
Using a functional expression strategy (Cl-dependent Na+ uptake in Xenopus oocytes), Gamba et al. (1993) isolated a cDNA clone from the urinary bladder of the winter flounder that codes for a Na+Cl symporter. This Na+Cl symporter is inhibited by a number of thiazide diuretics (but not by furosemide, acetazolamide, or an amiloride derivative), has 12 putative membrane-spanning domains, and its sequence is 47% identical to the cloned dogfish shark rectal gland Na+K+2Cl symporter. Subsequently, Gamba et al. (1994) cloned the rat and Mastroianni et al. (1996) cloned the human Na+Cl symporter. The Na+Cl symporter (called ENCC1 or TSC) is expressed predominantly in the kidney (Chang et al., 1996) and is localized to the apical membrane of DCT epithelial cells (Bachmann et al., 1995; Obermller et al., 1995; Plotkin et al., 1996). Expression of the Na+Cl symporter is regulated by aldosterone (Velzquez et al., 1996; Kim et al., 1998; Bostonjoglo et al., 1998). Mutations in the Na+Cl symporter cause a form of inherited hypokalemic alkalosis called Gitelman's syndrome (seeSimon and Lifton, 1998). Effects on Urinary Excretion As would be expected from their mechanism of action, inhibitors of Na+Cl symport increase Na+ and Cl excretion. However, thiazides are only moderately efficacious (i.e., maximum excretion of filtered load of Na+ is only 5%), since approximately 90% of the filtered Na+ load is reabsorbed before reaching the DCT. Some thiazide diuretics also are weak inhibitors of carbonic anhydrase, an effect that increases HCO3 and phosphate excretion and probably accounts for the weak proximal tubular effects of some thiazide diuretics. Like inhibitors of Na+K+2Cl symport, inhibitors of Na+Cl symport increase the excretion of K+ and titratable acid due to increased delivery of Na+ to the distal tubule. Acute administration of thiazides increases the excretion of uric acid. However, uric acid excretion is reduced following chronic administration by the same mechanisms discussed for loop diuretics. The acute effects of inhibitors of Na+Cl symport on Ca2+ excretion are variable; when administered chronically, thiazide diuretics decrease Ca2+ excretion. The mechanism is unknown but may involve increased proximal reabsorption due to volume depletion as well as direct effects of thiazides to increase Ca2+ reabsorption in the DCT. Thiazide diuretics may cause a mild magnesuria by a poorly understood mechanism, and there is increasing awareness that long-term use of thiazide diuretics may cause magnesium deficiency, particularly in the elderly (Martin and Milligan, 1987). Since inhibitors of Na+Cl symport inhibit transport in the cortical diluting segment, thiazide diuretics attenuate the ability of the kidney to excrete a dilute urine during water diuresis. However, since the DCT is not involved in the mechanism that generates a hypertonic medullary interstitium, thiazide diuretics do not alter the kidney's ability to concentrate urine during hydropenia. Effects on Renal Hemodynamics In general, inhibitors of Na+Cl symport do not affect RBF and only variably reduce GFR due to increases in intratubular pressure. Since thiazides act at a point past the macula densa, they have little or no influence on TGF. Other Actions Thiazide diuretics may inhibit phosphodiesterase, mitochondrial oxygen consumption, and renal uptake of fatty acids; however, these effects are not of clinical significance. Absorption and Elimination The relative potency, oral bioavailability, plasma half-life, and
route of elimination of inhibitors of Na+Cl symport
currently used in the Toxicity, Adverse Effects, Contraindications, Drug Interactions Thiazide diuretics rarely cause CNS (vertigo, headache, paresthesias,
xanthopsia, weakness), gastrointestinal (anorexia, nausea, vomiting, cramping,
diarrhea, constipation, cholecystitis, pancreatitis), hematological (blood
dyscrasias), and dermatological (photosensitivity, skin rashes) disorders.
The incidence of sexual dysfunction (i.e., erection problems) is
greater with Na+Cl symport inhibitors than with
several other antihypertensive agents ( Thiazide diuretics also decrease glucose tolerance, and latent diabetes mellitus may be unmasked during therapy. The mechanism of the reduced glucose tolerance is not completely understood but appears to involve reduced insulin secretion and alterations in glucose metabolism. Hyperglycemia may be related in some way to K+ depletion, in that hyperglycemia is reduced when K+ is given along with the diuretic (Tannen, 1985). Thiazide diuretics also may increase plasma levels of LDL cholesterol, total cholesterol, and total triglycerides. Thiazide diuretics are contraindicated in individuals who are hypersensitive to sulfonamides. With regard to drug interactions, thiazide diuretics may diminish the effects of anticoagulants, uricosuric agents used to treat gout, sulfonylureas, and insulin and may increase the effects of anesthetics, diazoxide, digitalis glycosides, lithium, loop diuretics, and vitamin D. The effectiveness of thiazide diuretics may be reduced by NSAIDs, bile acid sequestrants (reduced absorption of thiazides), and methenamines (alkalinization of urine may decrease effectiveness of thiazides). Amphotericin B and corticosteroids increase the risk of hypokalemia induced by thiazide diuretics. A potentially lethal drug interaction warranting special emphasis is that involving thiazide diuretics with quinidine (Roden, 1993). Prolongation of the QT-interval by quinidine can lead to the development of polymorphic ventricular tachycardia (torsades de pointes) due to triggered activity originating from early afterdepolarizations (seeChapter 35: Antiarrhythmic Drugs). Although usually self-limiting, torsades de pointes may deteriorate into fatal ventricular fibrillation. Hypokalemia increases the risk of quinidine-induced torsades de pointes, and thiazide diuretics cause hypokalemia. It is likely, therefore, that thiazide diureticinduced K+ depletion accounts for many cases of quinidine-induced torsades de pointes. Therapeutic Uses Thiazide diuretics are used for treatment of the edema associated with heart (congestive heart failure), liver (hepatic cirrhosis), and renal (nephrotic syndrome, chronic renal failure, acute glomerulonephritis) disease. With the exceptions of metolazone and indapamide, most thiazide diuretics are ineffective when GFR is <30 to 40 ml/min. Thiazide diuretics decrease blood pressure in hypertensive patients by increasing the slope of the renal pressurenatriuresis relationship (Saito and Kimura, 1996), and thiazide diuretics are widely used for the treatment of hypertension, either alone or in combination with other antihypertensive drugs (seeChapter 33: Antihypertensive Agents and the Drug Therapy of Hypertension). In this regard, thiazide diuretics are inexpensive, as efficacious as other classes of antihypertensive agents, and well tolerated. Thiazides can be administered once daily, do not require dose titration, and have few contraindications. Moreover, thiazides have additive or synergistic effects when combined with other classes of antihypertensive agents. Although thiazides may marginally increase the risk of sudden death (seeHoes and Grobbee, 1996) and renal cell carcinoma (Grossman et al., 1999), in general these agents are safe and reduce cardiovascular morbidity and mortality in hypertensive patients. Because the adverse effects of thiazides increase progressively in severity at doses higher than maximally effective antihypertensive doses, only low doses should be prescribed for hypertension (see Ramsey, 1999). Thiazide diuretics, which reduce urinary excretion of Ca2+, sometimes are employed to treat calcium nephrolithiasis and may be useful for the treatment of osteoporosis (seeChapter 62: Agents Affecting Calcification and Bone Turnover: Calcium, Phosphate, Parathyroid Hormone, Vitamin D, Calcitonin, and Other Compounds). Thiazide diuretics are also the mainstay for treatment of nephrogenic diabetes insipidus, reducing urine volume by up to 50%. The mechanism of this paradoxical effect remains unknown (Grnbeck et al., 1998). Since other halides are excreted by renal processes similar to those for Cl, thiazide diuretics may be useful for the management of Br intoxication. |
Inhibitors of Renal Epithelial Na+ Channels (K+-Sparing Diuretics)
|
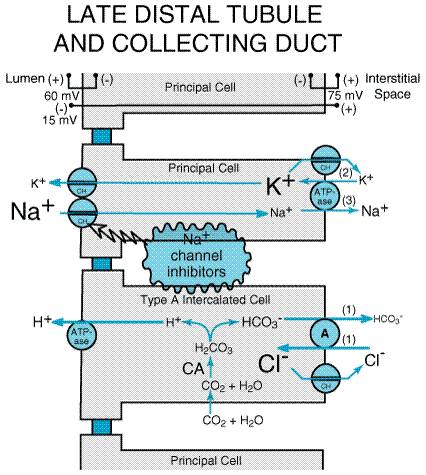
Triamterene DYRENIUM MAXZIDE ) and amiloride (MIDAMOR) are the only two drugs of this class in clinical use. Both drugs cause small increases in NaCl excretion and usually are employed for their antikaluretic actions to offset the effects of other diuretics that increase K+ excretion. Consequently, triamterene and amiloride, along with spironolactone (see next section), often are classified as potassium (K+)-sparing diuretics. Chemistry Amiloride is a pyrazinoylguanidine derivative, and triamterene is a pteridine (Table 296). Both drugs are organic bases and are transported by the organic base secretory mechanism in the proximal tubule. Mechanism and Site of Action Available data suggest that triamterene and amiloride have similar mechanisms of action. Of the two, amiloride has been studied much more extensively, so its mechanism of action is known with a higher degree of certainty. As illustrated in Figure 298, principal cells in the late distal tubule and collecting duct have in their luminal membranes a Na+ channel that provides a conductive pathway for the entry of Na+ into the cell down the electrochemical gradient created by the basolateral Na+ pump. The higher permeability of the luminal membrane for Na+ depolarizes the luminal membrane, but not the basolateral membrane, creating a lumen-negative transepithelial potential difference. This transepithelial voltage provides an important driving force for the secretion of K+ into the lumen via K+ channels (ROMK) in the luminal membrane. Carbonic anhydrase inhibitors, loop diuretics, and thiazide diuretics increase the delivery of Na+ to the late distal tubule and collecting duct, a situation that is often associated with increased K+ and H+ excretion. It is likely that the elevation in luminal Na+ concentration in the distal nephron induced by such diuretics augments depolarization of the luminal membrane and thereby enhances the lumen-negative VT, which facilitates K+ excretion. In addition to principal cells, the collecting duct also contains type A intercalated cells that mediate the secretion of H+ into the tubular lumen. Tubular acidification is driven by a luminal H+ATPase (proton pump), and this pump is aided by the lumen-negative transepithelial voltage. However, increased distal delivery of Na+ is not the only mechanism by which diuretics increase K+ and H+ excretion. Activation of the reninangiotensinaldosterone axis by diuretics also contributes to diuretic-induced K+ and H+ excretion by a mechanism explained in the section on mineralocorticoid antagonists.
Considerable evidence indicates that amiloride blocks Na+ channels in the luminal membrane of principal cells in the late distal tubule and collecting duct. This evidence includes data from epithelia of nonrenal origin (amphibian skin and toad bladder) (Garty and Palmer, 1997) as well as a number of electrophysiological studies in isolated mammalian collecting ducts (O'Neil and Boulpaep, 1979). Amiloride produces half-maximal inhibition at concentrations <1 mM and, depending on the study, amiloride may interact with Na+ in the channel either competitively or noncompetitively. It is important, however, to bear in mind that renal epithelial Na+ channels inhibited by this class of diuretics are not the same as voltage-gated Na+ channels found in many cell types (for instance, neurons and myocytes). Molecular cloning studies have revealed that the amiloride-sensitive
Na+ channel (called ENaC) consists of three subunits ( Effects on Urinary Excretion Since the late distal tubule and collecting duct have a limited capacity to reabsorb solutes, blockade of Na+ channels in this part of the nephron results in only a mild increase in the excretion rates of Na+ and Cl (approximately 2% of filtered load). Blockade of Na+ channels hyperpolarizes the luminal membrane, reducing the lumen-negative transepithelial voltage. Since the lumen-negative potential difference normally opposes cation reabsorption and facilitates cation secretion, attenuation of the lumen-negative voltage decreases the excretion rates of K+, H+, Ca2+, and Mg2+. Volume contraction may increase reabsorption of uric acid in the proximal tubule; hence, chronic administration of amiloride and triamterene may decrease uric acid excretion. Effects on Renal Hemodynamics Amiloride and triamterene have little or no effect on renal hemodynamics and do not alter TGF. Other Actions Amiloride, at concentrations higher than needed to elicit therapeutic effects, also blocks the Na+H+ and Na+Ca2+ antiporters and inhibits the Na+ pump. Absorption and Elimination The relative potency, oral bioavailability, plasma half-life, and route of elimination for amiloride and triamterene are listed in Table 296. Amiloride is eliminated predominantly by urinary excretion of intact drug. Triamterene is extensively metabolized to an active metabolite, 4-hydroxytriamterene sulfate, and this metabolite is excreted in the urine. The pharmacological activity of 4-hydroxytriamterene sulfate is comparable to that of the parent drug. Therefore, the toxicity of triamterene may be enhanced in both hepatic disease (decreased metabolism of triamterene) and renal failure (decreased urinary excretion of active metabolite). Toxicity, Adverse Effects, Contraindications, Drug Interactions The most dangerous adverse effect of Na+-channel inhibitors is hyperkalemia, which can be life-threatening. Consequently, amiloride and triamterene are contraindicated in patients with hyperkalemia as well as in patients at increased risk of developing hyperkalemia (e.g., patients with renal failure, patients receiving other K+-sparing diuretics, patients taking angiotensin converting enzyme inhibitors, or patients taking K+ supplements). Even NSAIDs can increase the likelihood of hyperkalemia in patients receiving Na+-channel inhibitors. Cirrhotic patients are prone to megaloblastosis because of folic acid deficiency, and triamterene, a weak folic acid antagonist, may increase the likelihood of this adverse event. Triamterene also can reduce glucose tolerance and induce photosensitization and has been associated with interstitial nephritis and renal stones. Both drugs can cause CNS, gastrointestinal, musculoskeletal, dermatological, and hematological adverse effects. The most common adverse effects of amiloride are nausea, vomiting, diarrhea, and headache; those of triamterene are nausea, vomiting, leg cramps, and dizziness. Therapeutic Uses Because of the mild natriuresis induced by Na+-channel inhibitors, these drugs seldom are used as sole agents in the treatment of edema or hypertension. Rather, their major utility is in combination with other diuretics. Coadministration of a Na+-channel inhibitor augments the diuretic and antihypertensive response to thiazide or loop diuretics. More importantly, the ability of Na+-channel inhibitors to reduce K+ excretion tends to offset the kaliuretic effects of thiazide and loop diuretics; consequently, the combination of a Na+-channel inhibitor with a thiazide or loop diuretic tends to result in normal values of plasma K+ (Hollenberg and Mickiewicz, 1989). Liddle's syndrome can be treated effectively with Na+-channel inhibitors. Aerosolized amiloride has been shown to improve mucociliary clearance in patients with cystic fibrosis (Zahaykevich, 1991). By inhibiting Na+ absorption from the surface of airway epithelial cells, amiloride augments hydration of respiratory secretions and thereby improves mucociliary clearance. Amiloride also is useful for lithium-induced nephrogenic diabetes insipidus because it blocks Li+ transport into the cells of the collecting tubules. |
Antagonists of Mineralocorticoid Receptors (Aldosterone Antagonists; K+-Sparing Diuretics)
|
Mineralocorticoids cause retention of salt
and water and increase the excretion of K+ and H+ by
binding to specific mineralocorticoid receptors. Kagawa et al. (1957)
observed that some spirolactones block the effects of mineralocorticoids;
this finding led to the synthesis of specific antagonists for the
mineralocorticoid receptor (MR). Spironolactone (ALDACTONE), a 17-spirolactone, is the only
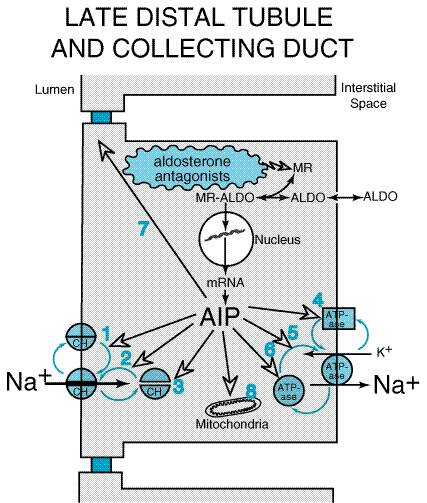
member of this class available in the Mechanism of Action Epithelial cells in the late distal tubule and collecting duct contain cytoplasmic MRs that have a high affinity for aldosterone. This receptor is a member of the superfamily of receptors for steroid hormones, thyroid hormones, vitamin D, and retinoids (seeChapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect). Aldosterone enters the epithelial cell from the basolateral membrane and binds to MRs; the MRaldosterone complex translocates to the nucleus, where it binds to specific sequences of DNA (hormone-responsive elements) and thereby regulates the expression of multiple gene products called aldosterone-induced proteins (AIPs). Figure 299 illustrates some of the proposed effects of AIPs, including: activation of 'silent' Na+ channels and 'silent' Na+ pumps that preexist in the cell membrane; alterations in the cycling of Na+ channels and Na+ pumps between the cytosol and cell membrane so that more channels and pumps are located in the membrane; increased expression of Na+ channels and Na+ pumps; changes in permeability of the tight junctions; and increased activity of enzymes in the mitochondria that are involved in ATP production. The precise mechanisms by which AIPs alter transport are incompletely understood. However, the net effect of AIPs is to increase Na+ conductance of the luminal membrane and sodium pump activity of the basolateral membrane. Consequently, transepithelial NaCl transport is enhanced and the lumen-negative transepithelial voltage is increased. The latter effect increases the driving force for secretion of K+ and H+ into the tubular lumen.
Drugs such as spironolactone competitively inhibit the binding of aldosterone to the MR (Marver et al., 1974). Unlike the MRaldosterone complex, the MRspironolactone complex is not able to induce the synthesis of AIPs. Since spironolactone and other drugs in this class block the biological effects of aldosterone, these agents also are referred to as aldosterone antagonists. Effects on Urinary Excretion The effects of spironolactone on urinary excretion are very similar to those induced by renal epithelial Na+-channel inhibitors. However, unlike that of the Na+-channel inhibitors, the clinical efficacy of spironolactone is a function of endogenous levels of aldosterone. The higher the levels of endogenous aldosterone, the greater the effects of spironolactone on urinary excretion. Effects on Renal Hemodynamics Spironolactone has little or no effect on renal hemodynamics and does not alter TGF. Other Actions High concentrations of spironolactone have been reported to interfere
with steroid biosynthesis by inhibiting 11 Absorption and Elimination Spironolactone is partially absorbed (approximately 65%), is
extensively metabolized (even during its first passage through the liver),
undergoes enterohepatic recirculation, is highly protein-bound, and has a
short half-life (approximately 1.6 hours). However, an active metabolite of
spironolactone, canrenone, has a half-life of approximately 16.5 hours, which
prolongs the biological effects of spironolactone. Although not available in
the Toxicity, Adverse Effects, Contraindications, Drug Interactions As with other K+-sparing diuretics, spironolactone may cause life-threatening hyperkalemia. Therefore, spironolactone is contraindicated in patients with hyperkalemia and in patients at increased risk of developing hyperkalemia, either because of disease or because of administration of other medications. Spironolactone also can induce metabolic acidosis in cirrhotic patients. Salicylates may reduce the tubular secretion of canrenone and decrease the diuretic efficacy of spironolactone, and spironolactone may alter the clearance of digitalis glycosides. Due to its steroid structure, spironolactone may cause gynecomastia, impotence, decreased libido, hirsutism, deepening of the voice, and menstrual irregularities. Spironolactone also may induce diarrhea, gastritis, gastric bleeding, and peptic ulcers (the drug is contraindicated in patients with peptic ulcers). CNS adverse effects include drowsiness, lethargy, ataxia, confusion, and headache. Spironolactone may cause skin rashes and, rarely, blood dyscrasias. Breast cancer has occurred in patients taking spironolactone chronically (cause and effect not established), and high doses of spironolactone have been associated with malignant tumors in rats. Whether or not therapeutic doses of spironolactone can induce malignancies remains an open question. Therapeutic Uses As with other K+-sparing diuretics, spironolactone often is coadministered with thiazide or loop diuretics in the treatment of edema and hypertension. Such combinations result in increased mobilization of edema fluid while causing lesser perturbations of K+ homeostasis. Spironolactone is particularly useful in the treatment of primary hyperaldosteronism (adrenal adenomas or bilateral adrenal hyperplasia) and of refractory edema associated with secondary aldosteronism (cardiac failure, hepatic cirrhosis, nephrotic syndrome, severe ascites). Spironolactone is considered the diuretic of choice in patients with hepatic cirrhosis. Pitt et al. (1999) have reported that spironolactone, when added to standard therapy, substantially reduces morbidity and mortality in patients with New York Heart Association (NYHA) class III and class IV heart failure (seeChapter 34: Pharmacological Treatment of Heart Failure). |
Mechanisms of Edema Formation and the Role of Diuretics in Clinical Medicine
|
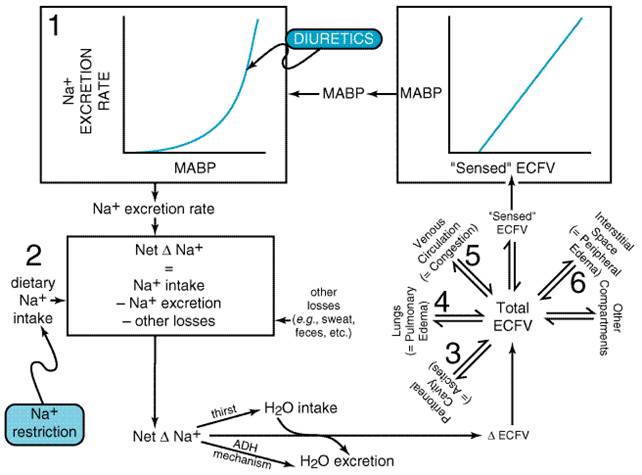
Mechanism of Edema Formation A complex set of interrelationships (Figure 2910) exists among the cardiovascular system, the kidneys, the CNS (Na+ appetite, thirst regulation), and the tissue capillary beds [distribution of extracellular fluid volume (ECFV)], so that perturbations at one of these sites can affect all of the remaining sites. A primary law of the kidney is that Na+ excretion is a steep function of mean arterial blood pressure (MABP) such that small increases in MABP cause marked increases in Na+ excretion (Guyton, 1991). Over any given time interval, the net change in total body Na+ (either positive or negative) is simply the dietary Na+ intake minus the urinary excretion rate minus other losses (e.g., sweating, fecal losses, vomiting). When a net positive Na+ balance occurs, the concentration of Na+ in the ECF will increase, stimulating water intake (thirst) and reducing urinary water output (via ADH release). Opposite changes occur during a net negative Na+ balance. Changes in water intake and output adjust ECFV concentration toward normal, thereby expanding or contracting total ECFV. Total ECFV is distributed among many body compartments; however, since the volume of extracellular fluid on the arterial side of the circulation pressurizes the arterial tree, it is this fraction of ECFV that determines MABP, and it is this fraction of ECFV that is 'sensed' by the cardiovascular system and kidneys. Since MABP is a major determinant of Na+ output, a closed loop is established (Figure 2910). This loop cycles until net Na+ accumulation is zero; i.e., in the long run, Na+ intake must equal Na+ loss.
The
above discussion implies that three fundamental types of perturbations
contribute to venous congestion and/or edema formation: (1) A shift to the
right in the renal pressurenatriuresis relationship (e.g., chronic
renal failure) causes reduced Na+ excretion for any level of MABP.
If all other factors remain constant, this would increase total body Na+,
ECFV, and MABP. The additional ECFV would be distributed throughout various
body compartments, according to the state of cardiac function and prevailing
Starling forces, and would predispose toward venous congestion and/or edema.
Even so, in the absence of any other predisposing factors for venous
congestion and/or edema, a rightward shift in the renal pressurenatriuresis
curve generally causes hypertension with only a slight (usually immeasurable)
increase in ECFV. As elucidated by Guyton and coworkers (Guyton, 1991), ECFV
expansion triggers the following series of events: expanded ECFV The Role of Diuretics in Clinical Medicine Another implication of the mechanisms illustrated in Figure 2910 is that three fundamental strategies exist for mobilizing edema fluidcorrect the underlying disease, restrict Na+ intake, or administer diuretics. The most desirable course of action would be to correct the primary disease; however, this is often impossible. For instance, the increased hepatic sinusoidal pressure in cirrhosis of the liver and the urinary loss of protein in nephrotic syndrome are due to structural alterations in the portal circulation and glomeruli, respectively, which may not be remediable. Restriction of Na+ intake is the favored nonpharmacological approach for the treatment of edema and hypertension and should usually be attempted; however, compliance is a major obstacle. Diuretics, therefore, remain the cornerstone for the treatment of edema or volume overload, particularly that due to congestive heart failure, ascites, chronic renal failure, and nephrotic syndrome. With regard to heart failure, diuretics reduce pulmonary edema and venous congestion, and it may be possible to manage mild cardiac failure with diuretics alone. However, most patients ultimately will require additional therapy with digitalis and/or angiotensin converting enzyme inhibitors (Chapter 34: Pharmacological Treatment of Heart Failure). Periodic administration of diuretics to cirrhotic patients with ascites may eliminate the necessity for or reduce the interval between paracenteses, adding to patient comfort and sparing protein reserves that are lost by paracenteses. Although diuretics can reduce the edema associated with chronic renal failure, increased doses of the more powerful loop diuretics may be required. In the nephrotic syndrome, the response to diuretics often is disappointing. Whether a patient should receive diuretics and, if so, what therapeutic regimen should be used (i.e., type of diuretic, route of administration, and speed of mobilization of edema fluid) depends on the clinical situation. Massive pulmonary edema in patients with acute left heart failure is a medical emergency requiring rapid, aggressive therapy including intravenous administration of a loop diuretic. In this setting, use of oral diuretics or diuretics with lesser efficacy is inappropriate. On the other hand, mild pulmonary and venous congestion associated with chronic heart failure is best treated with an oral loop diuretic, the dosage of which should be titrated carefully to maximize the benefit-to-risk ratio. In many situations, edema will not pose an immediate health risk. Even so, uncomfortable, oppressive, and/or disfiguring edema can greatly reduce quality of life, and the decision to treat will be based in part on quality-of-life issues. In such cases, only partial removal of edema fluid should be attempted, and the fluid should be mobilized slowly using a diuretic regimen that accomplishes the task with minimal perturbation of normal physiology. Brater (1998) has provided an algorithm for diuretic therapy (specific recommendations for drug, dose, route, and drug combinations) in patients with edema caused by renal, hepatic, or cardiac disorders. In many clinical situations, edema is not caused by an abnormal intake of Na+ or by an altered renal handling of Na+. Rather, edema is the result of altered Starling forces at the capillary beds, i.e., a 'Starling trap.' Use of diuretics in these clinical settings represents a judicious compromise between the edematous state and the hypovolemic state. In such conditions, reducing total ECFV with diuretics will decrease edema but also will cause depletion of 'sensed' ECFV, possibly leading to hypotension, malaise, and asthenia. Diuretic resistance refers to edema that is or has become refractory to a given diuretic. If diuretic resistance develops against a less efficacious diuretic, a more efficacious diuretic should be substituted, e.g., a loop diuretic for a thiazide. However, resistance to loop diuretics is not uncommon and can be due to several causes (Brater, 1985). NSAIDs block prostaglandin-mediated increases in RBF, resulting in resistance to loop diuretics. In chronic renal failure, a reduction in RBF decreases the delivery of diuretics to the kidney, and accumulation of endogenous organic acids competes with loop diuretics for transport at the proximal tubule. Consequently, the concentration of diuretic at the active site in the tubular lumen is diminished. In nephrotic syndrome, urinary protein binds diuretics and thereby limits the response. In hepatic cirrhosis or heart failure, the kidney may have a diminished responsiveness to diuretics because of increased proximal tubular Na+ reabsorption, leading to diminished delivery of Na+ to the distal nephron segments (Knauf and Mutschler, 1997). Faced with resistance to loop diuretics, the clinician has several options:
|
Prospectus
|
All currently available diuretics perturb K+ homeostasis. However, studies in animals have established that blockade of adenosine A1 receptors induces a brisk natriuresis without significantly increasing urinary K+ excretion (Kuan et al., 1993). Two clinical studies with FK453, a highly selective A1-receptor antagonist, confirm that blockade of A1 receptors induces natriuresis in human beings with minimal effects on K+ excretion (Balakrishnan et al., 1993; van Buren et al., 1993). The natriuretic mechanism of this novel class of diuretics has been partially elucidated (Takeda et al., 1993). Endogenous adenosine acts on A1 receptors in the proximal tubule to inhibit adenylyl cyclase. As cyclic AMP inhibits basolateral Na+HCO3 symport activity, a reduced level of cyclic AMP increases Na+HCO3 symport activity in the basolateral membrane. Blockade of A1 receptors prevents the inhibition of adenylyl cyclase by endogenous adenosine, increases epithelial cyclic AMP levels, and consequently decreases Na+HCO3 symport activity in the basolateral membrane of the proximal tubule. Because A1 receptors are involved in TGF, A1-receptor antagonists uncouple increased distal delivery of Na+ from activation of TGF (Wilcox et al., 1999). Other mechanisms, including an effect in the collecting tubules, contribute to the natriuretic response to A1-receptor antagonists; however, it is not known why this class of diuretics has little effect on K+ excretion. An A1-receptor antagonist is under clinical development for the treatment of edema due to heart failure. Recently, the water channels of the proximal tubule (aquaporin 1) and of the collecting duct (aquaporins 2, 3, and 4) were cloned and their functional characteristics examined (Agre et al., 1993; Fushimi et al., 1993; seeYamamoto and Sasaki, 1998). The cloning of these proteins represents an important step in our understanding of water homeostasis. Currently, there are no specific inhibitors of aquaporins; however, these proteins are important targets for the development of novel diuretics. Inhibition of water channels in the proximal tubule would greatly diminish the flux of water across proximal tubular epithelial cells, which would reduce luminal Na+ concentration to the point where Na+ reabsorption would cease. Therefore, inhibitors of proximal tubular aquaporin 1 may be useful natriuretic diuretics. On the other hand, inhibitors of collecting duct aquaporins 2, 3, and 4 would prevent water reabsorption in the collecting duct and therefore would be highly efficacious 'aquaretic' diuretics, i.e., diuretics with a predominant effect on water, rather than on Na+, excretion. Since aquaporin 2 is regulated by the vasopressin V2 receptor, another potential class of aquaretic diuretics would be nonpeptide, orally active V2-receptor antagonists. Exciting progress has been made, and this subject is developed in Chapter 30: Vasopressin and Other Agents Affecting the Renal Conservation of Water. Finally, apical K+ channels (ROMK) are another potential molecular target for the development of K+-sparing diuretics with high efficacy. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 10712
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved