| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Opioid Analgesics
Overview
|
Opioids have been the mainstay of pain treatment for thousands of years, and remain so today. Opioids exert their therapeutic effects by mimicking the action of endogenous opioid peptides at opioid receptors. Effects on both local neurons and intrinsic pain-modulating circuitry lead to analgesia, other therapeutic effects, and also to undesirable side effects. This chapter will provide the background necessary to understand the mechanisms of action and important pharmacological properties of clinically used opioids. First, the endogenous opioid system is discussed with a focus on the receptors and circuitry utilized by the opioids. A discussion of clinically used compounds follows, describing in detail their pharmacological properties and therapeutic uses. Routes of administration, pain treatment strategies, and current therapeutic guidelines also are presented. This information should provide a rational basis for understanding opioid actions, thereby reducing fear of opioid use and encouraging effective treatment of pain. |
Opioid Analgesics: Introduction
|
It is now well known that opioids such as heroin and morphine exert their effects by mimicking naturally occurring substances, termed endogenous opioid peptides or endorphins. Much now is known about the basic biology of the endogenous opioid system and its molecular and biochemical complexity, widespread anatomy, and diversity. The diverse functions of this system include the best-known sensory role, prominent in inhibiting responses to painful stimuli; a modulatory role in gastrointestinal, endocrine, and autonomic functions; an emotional role, evident in the powerful rewarding and addicting properties of opioids; and a cognitive role in the modulation of learning and memory. The endogenous opioid system is complex and subtle, with a great diversity in endogenous ligands (over a dozen), yet with only four major receptor types. This chapter presents key facts about the biochemical and functional nature of the opioid system. This information then is used to establish a basis for understanding the actions of clinically used opioid drugs and current strategies for pain treatment. Terminology The term opioid refers broadly to all compounds related to
opium. The word opium is derived from opos, the Greek word for
juice, the drug being derived from the juice of the opium poppy, Papaver
somniferum. Opiates are drugs derived from opium, and include the natural
products morphine, codeine, thebaine, and many semisynthetic congeners
derived from them. Endogenous opioid peptides are the naturally
occurring ligands for opioid receptors. The term endorphin is used
synonymously with endogenous opioid peptides, but also refers to a specific
endogenous opioid, History The first undisputed reference to opium is found in the writings of Theophrastus in the third century B.C. Arabian physicians were well versed in the uses of opium; Arabian traders introduced the drug to the Orient, where it was employed mainly for the control of dysenteries. During the Middle Ages, many of the uses of opium were appreciated. In 1680, Sydenham wrote: 'Among the remedies which it has pleased Almighty God to give to man to relieve his sufferings, none is so universal and so efficacious as opium.' Opium contains more than 20 distinct alkaloids. In 1806, Sertrner reported the isolation of a pure substance in opium that he named morphine, after Morpheus, the Greek god of dreams. The discovery of other alkaloids in opium quickly followedcodeine by Robiquet in 1832, and papaverine by Merck in 1848. By the middle of the nineteenth century, the use of pure alkaloids rather than crude opium preparations began to spread throughout the medical world. In addition to the remarkable beneficial effects of opioids, the toxic side effects and addictive potential of these drugs also have been known for centuries. These problems stimulated a search for potent, synthetic opioid analgesics free of addictive potential and other side effects. Unfortunately, all of the synthetic compounds that have been introduced into clinical use share the liabilities of classical opioids. However, the search for new opioid agonists led to the synthesis of opioid antagonists and compounds with mixed agonist/antagonist properties, which expanded therapeutic options and provided important tools for exploring mechanisms of opioid actions. Until the early 1970s, the endogenous opioid system was totally unknown. The actions of morphine, heroin, and other opioids as antinociceptive and addictive agents, while well described, often were studied in the context of interactions with other neurotransmitter systems, such as monoaminergic and cholinergic. Some investigators suggested the existence of a specific opioid receptor because of the unique structural requirements of opiate ligands (Beckett and Casy, 1954), but the presence of an opiate-like system in the brain remained unproven. A particularly misleading observation was that the administration of the opioid antagonist naloxone to a normal animal produced little effect, although the drug was effective in reversing or preventing the effects of exogenous opiates. The first physiological evidence suggesting an endogenous opioid system was the demonstration that analgesia produced by electrical stimulation of certain brain regions was reversed by naloxone (Akil et al., 1972; Akil et al., 1976). Pharmacological evidence for an opiate receptor also was building. In 1973, investigators in three laboratories demonstrated opiate binding sites in the brain (Pert and Snyder, 1973; Simon et al., 1973; Terenius, 1973). This was the first use of radioligand binding assays to demonstrate the presence of membrane-associated neurotransmitter receptors in the brain. Stimulation-produced analgesia, its naloxone reversibility, and the discovery of opioid receptors strongly pointed to the existence of endogenous opioids. In 1975, Hughes and associates identified an endogenous, opiate-like factor that they called enkephalin (from the head) (Hughes et al., 1975). Soon after, two more classes of endogenous opioid peptides were isolated, the dynorphins and endorphins. Details of these discoveries and the unique properties of the opioid peptides have been reviewed previously (Akil et al., 1984). Given the large number of endogenous ligands being discovered, it was
not surprising that multiple classes of opioid receptors also were found. The
concept of opioid-receptor multiplicity arose shortly after the initial
demonstration of opiate binding sites. Based on results of in vivo
studies in dogs, Martin and colleagues postulated the existence of multiple
types of opiate receptors (Martin et al., 1976). Receptor-binding
studies and subsequent cloning confirmed the existence of three main receptor
types, |
Endogenous Opioid Peptides
|
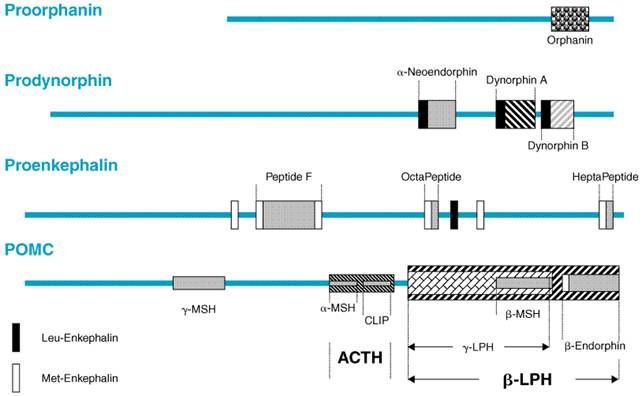
Three distinct families of classical opioid peptides have been identified: the enkephalins, endorphins, and dynorphins. Each family is derived from a distinct precursor polypeptide and has a characteristic anatomical distribution. These precursors, preproopiomelanocortin, preproenkephalin, and preprodynorphin, are encoded by three corresponding genes. Each precursor is subject to complex cleavages and posttranslational modifications resulting in the synthesis of multiple active peptides. The opioid peptides share the common amino-terminal sequence of Tyr-Gly-Gly-Phe- (Met or Leu), which has been called the 'opioid motif.' This motif is followed by various C-terminal extensions yielding peptides ranging from 5 to 31 residues (Table 231). The major opioid peptide derived from proopiomelanocortin (POMC) is
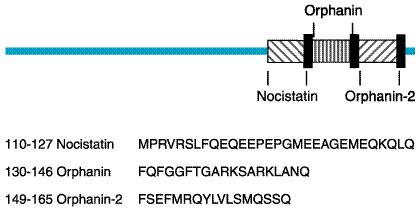
A novel endogenous opioid peptide was cloned in 1995 (Meunier et al., 1995; Reinscheid et al., 1995). This peptide has a significant sequence homology to dynorphin A, with an identical length of 17 amino acids, identical carboxy-terminal residues, and a slight modification of the amino-terminal opioid core (Phe-Gly-Gly-Phe instead of Tyr-Gly-Gly-Phe; see Table 231). The removal of this single hydroxyl group is sufficient to abolish interactions with the three classical opioid-peptide receptors. This peptide was called orphanin FQ (OFQ) by one group of investigators and nociceptin (N) by another, because it lowered pain threshold under certain conditions. The structure of the N/OFQ precursor (Figure 232) suggests that it may encode other biologically active peptides (Nothacker et al., 1996; Pan et al., 1996). Immediately downstream of N/OFQ is a 17-amino-acid peptide (orphanin-2), which also starts with phenylalanine and ends with glutamine but is otherwise distinct from N/OFQ, as well as a putative peptide upstream from N/OFQ, which may be liberated upon posttranslational processing (nocistatin). The N/OFQ system represents a new neuropeptide system with a high degree of sequence identity to the opioid peptides. However, the slight change in structure results in a profound alteration in function. N/OFQ has behavioral and pain modulatory properties distinct from those of the three classical opioid peptides (see below).
The anatomical distribution of POMC-producing cells is relatively limited within the CNS, occurring mainly in the arcuate nucleus and nucleus tractus solitarius. These neurons project widely to limbic and brainstem areas and to the spinal cord (Lewis et al., 1987). There also is evidence of POMC production in the spinal cord (Gutstein et al., 1992). The distribution of POMC corresponds to areas of the human brain where electrical stimulation can relieve pain (Pilcher et al., 1988). Peptides from POMC occur in both the pars intermedia and the pars distalis of the pituitary and also are contained in pancreatic islet cells. The peptides from prodynorphin and proenkephalin are distributed widely throughout the CNS and frequently are found together. Although each family of peptides usually is located in different groups of neurons, occasionally more than one family is expressed within the same neuron (Weihe et al., 1988). Of particular note, proenkephalin peptides are present in areas of the CNS that are presumed to be related to the perception of pain (e.g., laminae I and II of the spinal cord, the spinal trigeminal nucleus, and the periaqueductal gray), to the modulation of affective behavior (e.g., amygdala, hippocampus, locus ceruleus, and the cerebral cortex), to the modulation of motor control (caudate nucleus and globus pallidus), and the regulation of the autonomic nervous system (medulla oblongata) and neuroendocrinological functions (median eminence). Although there are a few long enkephalinergic fiber tracts, these peptides are contained primarily in interneurons with short axons. The peptides from proenkephalin also are found in the adrenal medulla and in nerve plexuses and exocrine glands of the stomach and intestine. The N/OFQ precursor has a unique anatomical distribution (Neal et al., 1999b). The distribution of this system suggests important roles in hippocampus, cortex, and numerous sensory sites. N/OFQ produces a complex behavioral profile, including effects on drug reward and reinforcement (Bertorelli et al., 2000; Devine et al., 1996a; Devine et al., 1996b), stress responsiveness (Devine et al., 2001; Koster et al., 1999), and learning and memory processes (Koster et al., 1999; Manabe et al., 1998). Studies of the effect of N/OFQ on pain sensitivity have produced conflicting results, which may be reconciled by data suggesting that the effects of N/OFQ on pain sensitivity depend on the underlying behavioral state of the animal (Pan et al., 2000) (see below). Analogous mechanisms also could explain some of the conflicting results with other physiological processes. However, more studies are needed before a general role can be ascribed to the N/OFQ system, including the investigation of other active peptides that may be derived from the N/OFQ precursor (Figure 232). Nocistatin has been tested behaviorally and found to produce effects opposite to those of N/OFQ (Okuda-Ashitaka et al., 1998). In sum, these findings, coupled with the extensive anatomy of the system, suggest that the N/OFQ precursor plays a complex role in the brain that is yet to be fully appreciated. Not all cells that make a given precursor polypeptide store and
release the same mixture of active opioid peptides, because of differential
processing secondary to variations in the cellular complement of peptidases
that produce and degrade the active opioid fragments (Akil et al.,
1984). In addition, processing of these peptides is altered by physiological
demands, leading to a different mix of peptides being released by the same
cell under different conditions. For example, chronic morphine treatment (Bronstein
et al., 1990) or stress (Akil et al., 1985) can alter the forms
of |
Opioid Receptors
|
Three classical opioid receptor types, The study of the biological functions of opioid receptors in vivo
was aided by the synthesis of selective antagonists and agonists. Among the
most commonly used antagonists are cyclic analogs of somatostatin such as
CTOP as Most of the clinically used opioids are relatively selective for There is little agreement regarding the exact classification of opioid
receptor subtypes. Pharmacological studies have suggested the existence of
multiple subtypes of each receptor. The complex literature on Molecular Studies of Opioid Receptors and Their Ligands For many years, the study of multiple opioid receptors greatly
profited from the availability of a rich array of natural and synthetic
ligands but was limited by the absence of opioid receptor clones. In 1992,
the mouse
It is possible that further cloning experiments may identify unique genes encoding opioid receptor subtypes. However, it has been suggested that, if multiple opioid receptor subtypes exist, they could be derived from a single gene, and multiple mechanisms might exist to achieve distinct pharmacological profiles. Two potential pathways to opioid receptor diversity are alternative splicing of receptor RNA and dimerization of receptor proteins. Alternative splicing of receptor heteronuclear RNA (e.g., exon
skipping and intron retention) is thought to play an important role in
producing in vivo diversity within many members of the GPCR
superfamily (Kilpatrick et al., 1999). Splice variants may exist
within each of the three opioid receptor families, and this alternative
splicing of receptor transcripts may be critical for the diversity of opioid
receptors. A technique widely used to identify potential sites of alternative
splicing is antisense oligodeoxynucleotide (ODN) mapping. The ability of
antisense ODNs to target specific regions of cDNA permits the systematic
evaluation of the contribution of individual exons to observed receptor
properties. Antisense ODN-targeting of exon 1 of the rat and mouse The interaction of two receptors to form a unique structure
(dimerization) also has been accorded an important role in regulating
receptor function. For example, dimerization of GABABR1 and GABABR2
subunits is required to form a functional GABAB receptor for
gamma-aminobutyric acid (e.g., Jones et al., 1998). Both cloned
Given the existence of four families of endogenous ligands and cloned
receptors, it seems reasonable to ask if there is a one-to-one correspondence
between them. Previous studies using brain homogenates demonstrated that an
orderly pattern of association between a set of opioid gene products and a
given receptor does not exist. Although proenkephalin products generally are
associated with Endomorphins The search for a high affinity/high selectivity endogenous ligand for
the Molecular Basis for Opioid Receptor Selectivity and Affinity Previous studies of other peptide receptors suggested that peptides and small molecules may bind to GPCRs differently. Mutagenesis studies of small ligand receptors (e.g., adrenergic and dopamine receptors) showed that charged amino acid residues in the transmembrane domains were important in receptor binding and activation (Strader et al., 1988; Mansour et al., 1992). This observation places the bound ligands within the receptor core formed by the transmembrane helices. On the other hand, studies with peptidergic receptors have demonstrated a critical role for extracellular loops in ligand recognition (Xie et al., 1990). All three classical opioid receptors appear to combine both properties: Charged residues located in transmembrane domains have been implicated in the high affinity binding of most opioid ligands, whether alkaloid or peptide (Surratt et al., 1994; Mansour et al., 1997). However, critical interactions of opioid peptides with the extracellular domains also have been shown. The opioid peptide Tyr-Gly-Gly-Phe core, sometimes termed the
'message,' appears to be necessary for interaction with the
receptor-binding pocket; however, peptide selectivity resides in the
carboxy-terminal extension beyond the tetrapeptide core, providing the
'address' (Schwyzer, 1986). When the carboxy-terminal domain is
long, it may interact with extracellular loops of the receptors, contributing
to selectivity in a way that cannot be achieved by the much smaller
alkaloids. Indeed, dynorphin A selectivity is dependent on the second
extracellular loop of the Results of the research discussed above imply that the alkaloids are
small enough to fit completely inside or near the mouth of the receptor core,
while peptides bind to the extracellular loops and simultaneously extend to
the receptor core to activate the common binding site. That one can truly
separate the binding of peptides and alkaloids is demonstrated most clearly
by a genetically engineered Opioid Receptor Signaling and Consequent Intracellular Events Coupling of Opioid Receptors to Second Messengers The Receptor Desensitization, Internalization, and Sequestration Following Chronic Exposure to Opioids Transient administration of opioids leads to a phenomenon termed acute
tolerance, whereas sustained administration leads to the development of
'classical' or chronic tolerance. Tolerance simply refers to
a decrease in effectiveness of a drug with its repeated administration.
Recent studies have focused on cellular mechanisms of acute tolerance.
Several investigators have shown that short-term desensitization probably
involves phosphorylation of the Like other GPCRs, both Traditionally, long-term tolerance has been thought to be associated
with increases in adenylyl cyclase activitya counter-regulation to the
decrease in cyclic AMP levels seen after acute opioid administration (Sharma et
al., 1977). Chronic treatment with An 'Apparent Paradox' A paradox in evaluating the function of endogenous opioid systems is that a host of endogenous ligands activate a small number of opioid receptors. This pattern is different from that of many other neurotransmitter systems, where a single ligand interacts with a large number of receptors having different structures and second messengers. Is this richness and complexity at the presynaptic level lost as multiple opioid ligands derived from different genes converge on only three receptors, or is this richness preserved through means yet to be discovered? One possibility is that all opioid receptors have not been revealed by molecular cloning. Other options include splice variants, dimerization, and posttranslational modification, as discussed previously. Even assuming that other receptors and variants will be found, the binding of many endogenous ligands to the three cloned classical receptors suggests a great deal of convergence. However, this convergence may be only apparent, since multiple mechanisms for achieving distinctive responses in the context of the biology described above may exist. Some issues to consider are as follows:
Understanding the complexity of endogenous opioid peptides and their patterns of interaction with multiple opioid receptors may help define the similarities and differences between the endogenous modulation of these systems and their activation by drugs. These insights could be important in devising treatment strategies that maximize beneficial properties of opioids (e.g., pain relief) while limiting their undesirable side effects such as tolerance, dependence, and addiction. |
Effects of Clinically Used Opioids
|
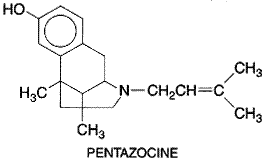
Morphine and most other clinically used
opioid agonists exert their effects through Mixed agonist-antagonist compounds were developed for clinical use with the hope that they would have less addictive potential and less respiratory depression than morphine and related drugs. In practice, however, it has turned out that for the same degree of analgesia, the same intensity of side effects will occur. (American Pain Society, 1999). A 'ceiling effect,' limiting the amount of analgesia attainable, often is seen with these drugs. Some mixed agonist-antagonist drugs, such as pentazocine and nalorphine, can produce severe psychotomimetic effects that are not reversible with naloxone (suggesting that these undesirable side effects are not mediated through classical opioid receptors). Also, pentazocine and nalorphine can precipitate withdrawal in opioid-tolerant patients. For these reasons, the clinical use of these mixed agonist-antagonist drugs is limited. Analgesia In human beings, morphine-like drugs produce analgesia, drowsiness, changes in mood, and mental clouding. A significant feature of the analgesia is that it occurs without loss of consciousness. When therapeutic doses of morphine are given to patients with pain, they report that the pain is less intense, less discomforting, or entirely gone; drowsiness commonly occurs. In addition to relief of distress, some patients experience euphoria. When morphine in the same dose is given to a normal, pain-free individual, the experience may be unpleasant. Nausea is common, and vomiting also may occur. There may be feelings of drowsiness, difficulty in mentation, apathy, and lessened physical activity. As the dose is increased, the subjective, analgesic, and toxic effects, including respiratory depression, become more pronounced. Morphine does not have anticonvulsant activity and usually does not cause slurred speech, emotional lability, or significant motor incoordination. The relief of pain by morphine-like opioids is relatively selective, in that other sensory modalities are not affected. Patients frequently report that the pain is still present, but that they feel more comfortable (see section on Therapeutic Uses of Opioid Analgesics). Continuous, dull pain is relieved more effectively than sharp, intermittent pain, but with sufficient amounts of opioid it is possible to relieve even the severe pain associated with renal or biliary colic. Any meaningful discussion of the action of analgesic agents must include some distinction between pain as a specific sensation, subserved by distinct neurophysiological structures, and pain as suffering (the original sensation plus the reactions evoked by the sensation). It is generally agreed that all types of painful experiences, whether produced experimentally or occurring clinically as a result of pathology, include both the original sensation and the reaction to that sensation. It also is important to distinguish between pain caused by stimulation of nociceptive receptors and transmitted over intact neural pathways (nociceptive pain) and pain that is caused by damage to neural structures, often involving neural supersensitivity (neuropathic pain). Although nociceptive pain usually is responsive to opioid analgesics, neuropathic pain typically responds poorly to opioid analgesics and may require higher doses of drug (McQuay, 1988). In clinical situations, pain cannot be terminated at will, and the meaning of the sensation and the distress it engenders are markedly affected by the individual's previous experiences and current expectations. In experimentally produced pain, measurements of the effects of morphine on pain threshold have not always been consistent; some workers find that opioids reliably elevate the threshold, while many others do not obtain consistent changes. In contrast, moderate doses of morphine-like analgesics are effective in relieving clinical pain and increasing the capacity to tolerate experimentally induced pain. Not only is the sensation of pain altered by opioid analgesics, but the affective response is changed as well. This latter effect is best assessed by asking patients with clinical pain about the degree of relief produced by the drug administered. When pain does not evoke its usual responses (anxiety, fear, panic, and suffering), a patient's ability to tolerate the pain may be markedly increased even when the capacity to perceive the sensation is relatively unaltered. It is clear, however, that alteration of the emotional reaction to painful stimuli is not the sole mechanism of analgesia. Intrathecal administration of opioids can produce profound segmental analgesia without causing significant alteration of motor or sensory function or subjective effects (Yaksh, 1988). Mechanisms and Sites of Opioid-Induced Analgesia While cellular and molecular studies of opioid receptors are invaluable in understanding their function, it is critical to place them in their anatomical and physiological context to fully understand the opioid system. Pain control by opioids needs to be considered in the context of brain circuits modulating analgesia and the functions of the various receptor types in these circuits. Excellent reviews of this topic are available (Fields et al., 1991; Harris, 1996). It has been well established that the analgesic effects of opioids
arise from their ability to inhibit directly the ascending transmission of
nociceptive information from the spinal cord dorsal horn and to activate pain
control circuits that descend from the midbrain, via the rostral
ventromedial medulla, to the spinal cord dorsal horn. Opioid peptides and
their receptors are found throughout these descending pain control circuits (Mansour
et al., 1995; Gutstein et al., 1998). The distribution of opioid receptors in descending pain control
circuits indicates substantial overlap between As mentioned above, there is significant opioid-receptor ligand binding, and little detectable receptor mRNA expression in the spinal cord dorsal horn, but high levels of opioid-receptor mRNA in DRG. This distribution might suggest that the actions of opioid-receptor agonists relevant to analgesia at the spinal level are predominantly presynaptic. At least one presynaptic mechanism with potential clinical significance is inhibition of spinal tachykinin signaling. It is well known that opioids decrease the pain-evoked release of tachykinins from primary afferent nociceptors (Jessell and Iversen, 1977; Yaksh et al., 1980). Recently, the significance of this effect has been questioned. Trafton et al. (1999) have demonstrated that at least 80% of tachykinin signaling in response to noxious stimulation remains intact after the intrathecal administration of large doses of opioids. These results suggest that, while opioid administration may reduce tachykinin release from primary afferent nociceptors, this reduction has little functional impact on the actions of tachykinins on postsynaptic pain-transmitting neurons. This implies that either tachykinins are not central to pain signaling and/or opioid-induced analgesia at the spinal level or that, contrary to the conclusions suggested by anatomical studies, presynaptic opioid actions may be of little analgesic significance. Just as important insights have been made into mechanisms of opioid-induced analgesia at the brainstem and spinal levels, progress also has been made in understanding forebrain mechanisms. It is well known that the actions of opioids in bulbospinal pathways are critical to their analgesic efficacy. The precise role of forebrain actions of opioids and whether or not these actions are independent of those in bulbospinal pathways are less well defined. It is clear that opioid actions in the forebrain contribute to analgesia, because decerebration prevents analgesia when rats are tested for pain sensitivity using the formalin test (Matthies and Franklin, 1992), and microinjection of opioids into several forebrain regions are analgesic in this test (Manning et al., 1994). However, because these manipulations frequently do not change the analgesic efficacy of opioids in measures of acute phasic nociception, such as the tailflick test, a distinction has been made between forebrain-dependent mechanisms for morphine-induced analgesia in the presence of tissue injury and bulbospinal mechanisms for this analgesia in the absence of tissue injury. In an important series of experiments, Manning and Mayer (1995a; 1995b) have shown that this distinction is not absolute. Analgesia induced by systemic administration of morphine in both the tailflick and formalin tests was disrupted either by lesioning or reversibly inactivating the central nucleus of the amygdala, demonstrating that opioid actions in the forebrain contribute to analgesia in measures of tissue damage as well as acute, phasic nociception. The involvement of the amygdala in analgesia is intriguing, as the amygdala has been implicated in the environmental activation of pain control circuits, and it projects extensively to brainstem regions involved in descending pain control (Manning and Mayer, 1995a; 1995b). Simultaneous administration of morphine at both spinal and supraspinal
sites results in synergy in analgesic response, with a tenfold reduction in
the total dose of morphine necessary to produce equivalent analgesia at
either site alone. The mechanisms responsible for spinal/supraspinal synergy
are readily distinguished from those involved with supraspinal analgesia (Pick
et al., 1992a). In addition to the well-described spinal/supraspinal
synergy, synergistic Opioids also can produce analgesia when administered peripherally. Opioid receptors are present on peripheral nerves (Fields et al., 1980) and will respond to peripherally applied opioids and locally released endogenous opioid compounds when 'up-regulated' during inflammatory pain states (Stein et al., 1991; Stein, 1993). During inflammation, immune cells capable of releasing endogenous opioids are present near sensory nerves, and a perineural defect allows opioids access to the nerves (Stein, 1993; Stein, 1995). It appears that this also may occur in neuropathic pain models (Kayser et al., 1995), perhaps because of the presence of immune cells near damaged nerves (Monaco et al., 1992) and perineural defects extant in these conditions. The Role of N/OFQ and Its Receptor in Pain Modulation N/OFQ mRNA and peptide are present throughout descending pain control circuits. For instance, N/OFQ-containing neurons are present in the PAG, the median raphe, throughout the RVM, and in the superficial dorsal horn (Neal et al., 1999b). This distribution overlaps with that of opioid peptides, but the extent of colocalization remains unclear. N/OFQ-receptor ligand binding and mRNA are seen in the PAG, median raphe, and RVM (Neal et al., 1999a). Spinally, there is stronger N/OFQ-receptor mRNA expression in the ventral horn than in the dorsal horn, but higher levels of ligand binding in the dorsal horn. There also are high N/OFQ-receptor mRNA levels in the DRG. Despite clear anatomical evidence for a role of the N/OFQ system in pain modulation, its function remains unclear. Targeted disruption of the N/OFQ receptor in mice had little effect on basal pain sensitivity in several measures, whereas targeted disruption of the N/OFQ precursor consistently elevated basal responses in the tailflick test, suggesting an important role for N/OFQ in regulating basal pain sensitivity (Nishi et al., 1997; Koster et al., 1999). Intratheca1 injections of N/OFQ have been shown to be analgesic (Yamamoto et al., 1997; Xu et al., 1996); however, supraspinal administration has produced either hyperalgesia, antiopioid effects, or a biphasic hyperalgesic/analgesic response (Rossi et al., 1996b, Rossi et al., 1997; Grisel et al., 1996). These conflicting findings may be explained in part by a study in which it was shown that N/OFQ inhibits both pain-facilitating and analgesia-facilitating neurons in the RVM (Pan et al., 2000). Activation of endogenous analgesic circuitry was blocked by administration of N/OFQ. If the animal was hyperalgesic, the enhanced pain sensitivity also was blocked by N/OFQ. Thus, the effects of N/OFQ on pain responses appear to depend on the preexisting state of pain in the animal. Mood Alterations and Rewarding Properties The mechanisms by which opioids produce euphoria, tranquility, and other alterations of mood (including rewarding properties) are not entirely clear. However, the neural systems that mediate opioid reinforcement are distinct from those involved in physical dependence and analgesia (Koob and Bloom, 1988). Behavioral and pharmacological evidence points to the role of dopaminergic pathways, particularly involving the nucleus accumbens (NAcc), in drug-induced reward. There is ample evidence for interactions between opioids and dopamine in mediating opioid-induced reward. A full appreciation of mechanisms of drug-induced reward requires a more complete understanding of the NAcc and related structures at the anatomical level as well as a careful examination of the interface between the opioid system and dopamine receptors. The NAcc, portions of the olfactory tubercle, and the ventral and medial portions of the caudate-putamen constitute an area referred to as the ventral striatum (Heimer et al., 1982). The ventral striatum is implicated in motivation and affect (limbic functions), while the dorsal striatum is involved in sensorimotor and cognitive functions (Willner et al., 1991). Both the dorsal and ventral striatum are heterogeneous structures that can be subdivided into distinct compartments. In the middle and caudal third of the NAcc, the characteristic distribution of neuroactive substances results in two unique compartments termed the core and the shell (Zahm and Heimer, 1988; Heimer et al., 1991). It is important to note that other reward-relevant brain regions (e.g., the lateral hypothalamus and medial prefrontal cortex) implicated with a variety of abused drugs are connected reciprocally to the shell of the NAcc. Thus, the shell of the NAcc is the site that may be involved directly in the emotional and motivational aspects of drug-induced reward. Prodynorphin- and proenkephalin-derived opioid peptides are expressed
primarily in output neurons of the striatum and NAcc. All three opioid
receptor types are present in the NAcc (Mansour et al., 1988) and are
thought to mediate, at least in part, the motivational effects of opiate
drugs. Selective The locus ceruleus (LC) contains both noradrenergic neurons and high concentrations of opioid receptors and is postulated to play a critical role in feelings of alarm, panic, fear, and anxiety. Neural activity in the LC is inhibited by both exogenous opioids and endogenous opioid-like peptides. Other CNS Effects While opioids are used clinically primarily for their pain-relieving properties, they produce a host of other effects. This is not surprising in view of the wide distribution of opioids and their receptors, both in the brain and in the periphery. A brief summary of some of these effects is presented below. High doses of opioids can produce muscular rigidity in human beings. Chest wall rigidity severe enough to compromise respiration is not uncommon during anesthesia with fentanyl, alfentanil, remifentanil, and sufentanil (see Monk et al., 1988). Opioids and endogenous peptides cause catalepsy, circling, and stereotypical behavior in rats and other animals. Effects on the Hypothalamus Opioids alter the equilibrium point of the hypothalamic heat-regulatory mechanisms, such that body temperature usually falls slightly. However, chronic high dosage may increase body temperature (see Martin, 1983). Neuroendocrine Effects Morphine acts in the hypothalamus to inhibit the release of
gonadotropin-releasing hormone (GnRH) and corticotropin-releasing factor
(CRF), thus decreasing circulating concentrations of luteinizing hormone
(LH), follicle-stimulating hormone (FSH), ACTH, and The administration of Although Miosis Morphine and most Convulsions In animals, high doses of morphine and related opioids produce
convulsions. Several mechanisms appear to be involved, and different types of
opioids produce seizures with different characteristics. Morphine-like drugs
excite certain groups of neurons, especially hippocampal pyramidal cells;
these excitatory effects probably result from inhibition of the release of
GABA by interneurons (see McGinty and Friedman, 1988). Selective Respiration Morphine-like opioids depress respiration, at least in part by virtue of a direct effect on the brainstem respiratory centers. The respiratory depression is discernible even with doses too small to disturb consciousness and increases progressively as the dose is increased. In human beings, death from morphine poisoning is nearly always due to respiratory arrest. Therapeutic doses of morphine in human beings depress all phases of respiratory activity (rate, minute volume, and tidal exchange) and also may produce irregular and periodic breathing. The diminished respiratory volume is due primarily to a slower rate of breathing, and with toxic amounts the rate may fall to 3 or 4 breaths per minute. Although effects on respiration are readily demonstrated, clinically significant respiratory depression rarely occurs with standard morphine doses in the absence of underlying pulmonary dysfunction. However, the combination of opioids with other medications, such as general anesthetics, tranquilizers, alcohol, or sedative-hypnotics, may present a greater risk of respiratory depression. Maximal respiratory depression occurs within 5 to 10 minutes after intravenous administration of morphine or within 30 or 90 minutes following intramuscular or subcutaneous administration, respectively. Maximal respiratory depressant effects occur more rapidly with more lipid-soluble agents. Following therapeutic doses, respiratory minute volume may be reduced for as long as 4 to 5 hours. The primary mechanism of respiratory depression by opioids involves a reduction in the responsiveness of the brainstem respiratory centers to carbon dioxide. Opioids also depress the pontine and medullary centers involved in regulating respiratory rhythmicity and the responsiveness of medullary respiratory centers to electrical stimulation (see Martin, 1983). Hypoxic stimulation of the chemoreceptors still may be effective when
opioids have decreased the responsiveness to CO2, and the
inhalation of O2 may thus produce apnea. After large doses of morphine
or other Because of the accumulation of CO2, respiratory rate and sometimes even minute volume can be unreliable indicators of the degree of respiratory depression that has been produced by morphine. Natural sleep also produces a decrease in the sensitivity of the medullary center to CO2, and the effects of morphine and sleep are additive. Numerous studies have compared morphine and morphine-like opioids with
respect to their ratios of analgesic to respiratory-depressant activities.
Most studies have found that, when equianalgesic doses are used, the degree
of respiratory depression observed with morphine-like opioids is not
significantly different from that seen with morphine. Severe respiratory
depression is less likely after the administration of large doses of
selective Cough Morphine and related opioids also depress the cough reflex, at least in part by a direct effect on a cough center in the medulla. There is, however, no obligatory relationship between depression of respiration and depression of coughing, and effective antitussive agents are available that do not depress respiration (see below). Suppression of cough by such agents appears to involve receptors in the medulla that are less sensitive to naloxone than are those responsible for analgesia. Nauseant and Emetic Effects Nausea and vomiting produced by morphine-like drugs are unpleasant side effects caused by direct stimulation of the chemoreceptor trigger zone for emesis, in the area postrema of the medulla. Certain individuals never vomit after morphine, whereas others do so each time the drug is administered. Nausea and vomiting are relatively uncommon in recumbent patients
given therapeutic doses of morphine, but nausea occurs in approximately 40%
and vomiting in 15% of ambulatory patients given 15 mg of the drug
subcutaneously. This suggests that a vestibular component also is operative.
Indeed, the nauseant and emetic effects of morphine are markedly enhanced by
vestibular stimulation, and morphine and related synthetic analgesics produce
an increase in vestibular sensitivity. All clinically useful Cardiovascular System In the supine patient, therapeutic doses of morphine-like opioids have no major effect on blood pressure or cardiac rate and rhythm. Such doses do produce peripheral vasodilation, reduced peripheral resistance, and an inhibition of baroreceptor reflexes. Therefore, when supine patients assume the head-up position, orthostatic hypotension and fainting may occur. The peripheral arteriolar and venous dilation produced by morphine involves several mechanisms. Morphine and some other opioids provoke release of histamine, which sometimes plays a large role in the hypotension. However, vasodilation is usually only partially blocked by H1 antagonists, but it is effectively reversed by naloxone. Morphine also blunts the reflex vasoconstriction caused by increased PCO Effects on the myocardium are not significant in normal individuals. In patients with coronary artery disease but no acute medical problems, 8 to 15 mg of morphine administered intravenously produces a decrease in oxygen consumption, left ventricular end-diastolic pressure, and cardiac work; effects on cardiac index are usually slight (Sethna et al., 1982). In patients with acute myocardial infarction, the cardiovascular responses to morphine may be more variable than in normal subjects, and the magnitude of changes (e.g., the decrease in blood pressure) may be more pronounced (see Roth et al., 1988). Morphine may exert its well-known therapeutic effect in the treatment of angina pectoris and acute myocardial infarction by decreasing preload, inotropy, and chronotropy, thus favorably altering determinants of myocardial oxygen consumption and helping to relieve ischemia. It is not clear whether the analgesic properties of morphine in this situation are due to the reversal of acidosis that may stimulate local acid-sensing ion channels (Benson et al., 1999; McCleskey and Gold, 1999) or to a direct analgesic effect on nociceptive afferents from the heart. When administered prior to experimental ischemia, morphine has been
shown to produce cardioprotective effects. Morphine can mimic the phenomenon
of ischemic preconditioning, where a short ischemic episode paradoxically
protects the heart against further ischemia. This effect appears to be
mediated through Very large doses of morphine can be used to produce anesthesia;
however, decreased peripheral resistance and blood pressure are troublesome. Fentanyl
and sufentanil, which are potent and selective Morphine-like opioids should be used with caution in patients who have a decreased blood volume, since these agents can aggravate hypovolemic shock. Morphine should be used with great care in patients with cor pulmonale, since deaths following ordinary therapeutic doses have been reported. The concurrent use of certain phenothiazines may increase the risk of morphine-induced hypotension. Cerebral circulation is not directly affected by therapeutic doses of morphine. However, opioid-induced respiratory depression and CO2 retention can result in cerebral vasodilation and an increase in cerebrospinal fluid pressure; the pressure increase does not occur when PCO is maintained at normal levels by artificial ventilation. Gastrointestinal Tract Stomach Morphine and other Small Intestine Morphine diminishes biliary, pancreatic, and intestinal secretions (Dooley et al., 1988) and delays digestion of food in the small intestine. Resting tone is increased, and periodic spasms are observed. The amplitude of the nonpropulsive type of rhythmic, segmental contractions usually is enhanced, but propulsive contractions are markedly decreased. The upper part of the small intestine, particularly the duodenum, is affected more than the ileum. A period of relative atony may follow the hypertonicity. Water is absorbed more completely because of the delayed passage of bowel contents, and intestinal secretion is decreased; this increases the viscosity of the bowel contents. In the presence of intestinal hypersecretion that may be associated
with diarrhea, morphine-like drugs inhibit the transfer of fluid and
electrolytes into the lumen by naloxone-sensitive actions on the intestinal
mucosa and within the CNS. Enterocytes may possess opioid receptors, but this
hypothesis is controversial. However, it is clear that opioids exert
important effects on the submucosal plexus that lead to a decrease in the
basal secretion by enterocytes and inhibition of the stimulatory effects of
acetylcholine, prostaglandin E2, and vasoactive intestinal
peptide. The effects of opioids initiated either in the CNS or the submucosal
plexus may be mediated in large part by the release of norepinephrine and
stimulation of Large Intestine Propulsive peristaltic waves in the colon are diminished or abolished after administration of morphine, and tone is increased to the point of spasm. The resulting delay in the passage of bowel contents causes considerable desiccation of the feces, which, in turn, retards their advance through the colon. The amplitude of the nonpropulsive type of rhythmic contractions of the colon usually is enhanced. The tone of the anal sphincter is greatly augmented, and reflex relaxation in response to rectal distension is reduced. These actions, combined with inattention to the normal sensory stimuli for defecation reflex due to the central actions of the drug, contribute to morphine-induced constipation. Mechanism of Action on the Bowel The usual gastrointestinal effects of morphine primarily are mediated
by Biliary Tract After the subcutaneous injection of 10 mg of morphine sulfate, the sphincter of Oddi constricts and the pressure in the common bile duct may rise more than tenfold within 15 minutes; this effect may persist for 2 hours or more. Fluid pressure also may increase in the gallbladder and produce symptoms that may vary from epigastric distress to typical biliary colic. Some patients with biliary colic may experience exacerbation rather than relief of pain when given these drugs. Spasm of the sphincter of Oddi is probably responsible for elevations of plasma amylase and lipase that are sometimes found after patients are given morphine. Atropine only partially prevents morphine-induced biliary spasm, but opioid antagonists prevent or relieve it. Nitroglycerin (0.6 to 1.2 mg) administered sublingually also decreases the elevated intrabiliary pressure (see Staritz, 1988). Other Smooth Muscle Ureter and Urinary Bladder Therapeutic doses of morphine may increase the tone and amplitude of contractions of the ureter, although the response is variable. When the antidiuretic effects of the drug are prominent and urine flow decreases, the ureter may become quiescent. Morphine inhibits the urinary voiding reflex, and both the tone of the
external sphincter and the volume of the bladder are increased;
catheterization is sometimes required following therapeutic doses of
morphine. Stimulation of either Uterus If the uterus has been made hyperactive by oxytocics, morphine tends to restore tone, frequency, and the amplitude of contractions to normal. Parenteral administration of opioids within 2 to 4 hours of delivery may lead to transient respiratory depression in the neonate due to transplacental passage of opioids. This may be treated readily with naloxone. Skin Therapeutic doses of morphine cause dilation of cutaneous blood vessels. The skin of the face, neck, and upper thorax frequently becomes flushed. These changes may be due in part to the release of histamine and may be responsible for the sweating and some of the pruritus that occasionally follow the systemic administration of morphine (see below). Histamine release probably accounts for the urticaria commonly seen at the site of injection; this is not mediated by opioid receptors and is not blocked by naloxone. It is seen with morphine and meperidine, but not with oxymorphone, methadone, fentanyl, or sufentanil (see Duthie and Nimmo, 1987). Pruritus is a common and potentially disabling complication of opioid use. It can be caused by intraspinal and systemic injections of opioids, but it appears to be more intense after intraspinal administration (Ballantyne et al., 1988). The effect appears to be mediated in large part by dorsal horn neurons and is reversible by naloxone (Thomas et al., 1992). An intriguing report suggested that systemic morphine could partially inhibit pruritus caused by intraspinal administration of morphine, implying the existence of an opioid-mediated, itch-inhibition system, possibly supraspinal in origin (Thomas et al., 1993). Immune System The effects of opioids on the immune system are complex. Opioids have
been shown to modulate immune function by direct effects on cells of the
immune system and indirectly via centrally mediated neuronal
mechanisms (Sharp and Yaksh, 1997). It appears that acute, central
immunomodulatory effects of opioids may be mediated by activation of the
sympathetic nervous system, whereas the chronic effects of opioids may
involve modulation of hypothalamic-pituitary-adrenal (HPA) axis function (Mellon
and Bayer, 1998). Direct effects on immune cells may involve unique and as
yet incompletely characterized variants of the classical neuronal opioid
receptors, with The overall effects of opioids on immune function appear to be
suppressive; increased susceptibility to infection and tumor spread have been
observed in experimental settings. Infusion of the Tolerance and Physical Dependence The development of tolerance and physical dependence with repeated use is a characteristic feature of all the opioid drugs. Tolerance to the effect of opioids or other drugs simply means that, over time, the drug loses its effectiveness and an increased dose is required to produce the same physiological response. Dependence refers to a complex and poorly understood set of changes in the homeostasis of an organism that cause a disturbance of the homeostatic set point of the organism if the drug is stopped. This disturbance often is revealed when administration of an opioid is abruptly stopped, resulting in withdrawal. Addiction is a behavioral pattern characterized by compulsive use of a drug and overwhelming involvement with its procurement and use. Tolerance and dependence are physiological responses seen in all patients and are not predictors of addiction (see Chapter 24: Drug Addiction and Drug Abuse). These processes appear to be quite distinct. For example, cancer pain often requires prolonged treatment with high doses of opioids, leading to tolerance and dependence. Yet, abuse in this setting is very unusual (Foley, 1993). Neither the presence of tolerance and dependence nor the fear that they may develop should ever interfere with the appropriate use of opioids. Opioids can be discontinued in dependent patients once the need for analgesics is gone without subjecting them to withdrawal (see Chapter 24: Drug Addiction and Drug Abuse). Clinically, the dose can be decreased by 10% to 20% every other day and eventually stopped without signs and symptoms of withdrawal. In vivo
studies in animal models demonstrate the importance of other
neurotransmitters and their interactions with opioid pathways in the development
of tolerance to morphine. Blockade of glutamate actions by NMDA (N-methyl-D-aspartate)-receptor antagonists
blocks morphine tolerance (Trujillo and Akil, 1997). Since NMDA antagonists
have no effect on the potency of morphine in naive animals, their effect
cannot be attributed to potentiation of opioid actions. Interestingly, the
clinically used antitussive dextromethorphan (see Dextromethorphan)
has been shown to function as an NMDA antagonist. In animals, it can
attenuate opioid tolerance development and reverse established tolerance (Elliott
et al., 1994). Nitric oxide production, possibly induced by
NMDA-receptor activation, also has been implicated in tolerance, as
inhibition of nitric oxide synthase (NOS) also blocks morphine tolerance development
(Kolesnikov et al., 1993). Administering NOS inhibitors to
morphine-tolerant animals also may reverse tolerance in certain
circumstances. Although the NMDA antagonists and nitric oxide synthase
inhibitors are effective against tolerance to morphine and |
Morphine and Related Opioid Agonists
|
There are now many compounds with
pharmacological properties similar to those of morphine, yet morphine remains
the standard against which new analgesics are measured. However, responses of
an individual patient may vary dramatically with different Source and Composition of Opium Because the laboratory synthesis of morphine is difficult, the drug is still obtained from opium or extracted from poppy straw. Opium is obtained from the unripe seed capsules of the poppy plant, Papaver somniferum. The milky juice is dried and powdered to make powdered opium, which contains a number of alkaloids. Only a fewmorphine, codeine, and papaverinehave clinical usefulness. These alkaloids can be divided into two distinct chemical classes, phenanthrenes and benzylisoquinolines. The principal phenanthrenes are morphine (10% of opium), codeine (0.5%), and thebaine (0.2%). The principal benzylisoquinolines are papaverine (1.0%), which is a smooth muscle relaxant (see the seventh and earlier editions of this book), and noscapine (6.0%). Chemistry of Morphine and Related Opioids The structure of morphine is shown in Table 235. Many semisynthetic
derivatives are made by relatively simple modifications of morphine or
thebaine. Codeine is methylmorphine, the methyl substitution being on the
phenolic hydroxyl group. Thebaine differs from morphine only in that both
hydroxyl groups are methylated and that the ring has two double bonds ( Structure-Activity Relationship of the Morphine-Like Opioids In addition to morphine, codeine, and the semisynthetic derivatives of
the natural opium alkaloids, a number of other structurally distinct chemical
classes of drugs have pharmacological actions similar to those of morphine.
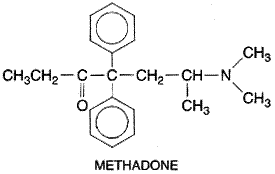
Clinically useful compounds include the morphinans, benzomorphans,
methadones, phenylpiperidines, and propionanilides. Although the
two-dimensional representations of these chemically diverse compounds appear
to be quite different, molecular models show certain common characteristics;
these are indicated by the heavy lines in the structure of morphine shown in Table
235. Among the important properties of the opioids that can be altered by
structural modification are their affinities for various species of opioid
receptors, their activities as agonists versus antagonists, their
lipid solubilities, and their resistance to metabolic breakdown. For example,
blockade of the phenolic hydroxyl at position 3, as in codeine and heroin,
drastically reduces binding to Absorption, Distribution, Fate, and Excretion Absorption In general, the opioids are readily absorbed from the gastrointestinal tract; absorption through the rectal mucosa is adequate, and a few agents (e.g., morphine, hydromorphone) are available in suppositories. The more lipophilic opioids also are readily absorbed through the nasal or buccal mucosa (Weinberg et al., 1988). Those with the greatest lipid solubility also can be absorbed transdermally (Portenoy et al., 1993). Opioids are absorbed readily after subcutaneous or intramuscular injection and can adequately penetrate the spinal cord following epidural or intrathecal administration (also see section on Alternative Routes of Administration). Small amounts of morphine introduced epidurally or intrathecally into the spinal canal can produce profound analgesia that may last 12 to 24 hours. However, due to the hydrophilic nature of morphine, there is rostral spread of the drug in spinal fluid, and side effects, especially respiratory depression, can emerge up to 24 hours later as the opioid reaches supraspinal respiratory control centers. With highly lipophilic agents such as hydromorphone or fentanyl, rapid absorption by spinal neural tissues produces very localized effects and segmental analgesia. The duration of action is shorter because of distribution of the drug in the systemic circulation, and the severity of respiratory depression may be more directly proportional to its concentration in plasma, due to a lesser degree of rostral spread (Gustafsson and Wiesenfeld-Hallin, 1988). However, patients receiving epidural or intrathecal fentanyl still should be monitored for respiratory depression. With most opioids, including morphine, the effect of a given dose is less after oral than after parenteral administration, due to variable but significant first-pass metabolism in the liver. For example, the bioavailability of oral preparations of morphine is only about 25%. The shape of the time-effect curve also varies with the route of administration, so that the duration of action is often somewhat longer with the oral route. If adjustment is made for variability of first-pass metabolism and clearance, it is possible to achieve adequate relief of pain by the oral administration of morphine. Satisfactory analgesia in cancer patients has been associated with a very broad range of steady-state concentrations of morphine in plasma (16 to 364 ng/ml; Neumann et al., 1982). When morphine and most opioids are given intravenously, they act promptly. However, the more lipid-soluble compounds act more rapidly than morphine after subcutaneous administration because of differences in the rates of absorption and entry into the CNS. Compared with other more lipid-soluble opioids such as codeine, heroin, and methadone, morphine crosses the bloodbrain barrier at a considerably lower rate. Distribution and Fate When therapeutic concentrations of morphine are present in plasma, about one-third of the drug is protein bound. Morphine itself does not persist in tissues, and 24 hours after the last dose tissue concentrations are low. The major pathway for the metabolism of morphine is conjugation with glucuronic acid. The two major metabolites formed are morphine-6-glucuronide and morphine-3-glucuronide. Small amounts of morphine 3,6, diglucuronide also may be formed. Although the 3- and 6-glucuronides are quite polar, both can cross the bloodbrain barrier to exert significant clinical effects (Christup, 1997). Morphine-6-glucuronide has pharmacological actions indistinguishable from those of morphine. Morphine-6-glucuronide given systemically is approximately twice as potent as morphine in animal models (Paul et al., 1989) and in human beings (Osborne et al., 1988). With chronic administration, it accounts for a significant portion of morphine's analgesic actions (Osborne et al., 1988; Osborne et al., 1990; Portenoy et al., 1991; Portenoy et al., 1992). Indeed, with chronic oral dosing, the blood levels of morphine-6-glucuronide typically exceed those of morphine. Given its greater potency as well as its higher concentrations, morphine-6-glucuronide may be responsible for most of morphine's analgesic activity in patients receiving chronic oral morphine. Morphine-6-glucuronide is excreted by the kidney. In renal failure, the levels of morphine-6-glucuronide can accumulate, perhaps explaining morphine's potency and long duration in patients with compromised renal function. In young adults, the half-life of morphine is about 2 hours; the half-life of morphine-6-glucuronide is somewhat longer. Children achieve adult renal function values by 6 months of age. In elderly patients, lower morphine doses are recommended, based on its smaller volume of distribution (Owen et al., 1983) and the general decline in renal function in the elderly. The 3-glucuronide, also an important metabolite of morphine (Milne et al., 1996), has little affinity for opioid receptors but may contribute to excitatory effects of morphine (Smith, 2000). Some investigators also have shown that morphine-3-glucuronide can antagonize morphine-induced analgesia (Smith et al., 1990), but this finding is not universal (Christup, 1997). Morphine also is metabolized by other pathways. N-demethylation to normorphine is a minor metabolic pathway in human beings but is more prominent in rodents (Yeh et al., 1977). N-dealkylation is important in the metabolism of some congeners of morphine. Excretion Very little morphine is excreted unchanged. It is eliminated by glomerular filtration, primarily as morphine-3-glucuronide; 90% of the total excretion takes place during the first day. Enterohepatic circulation of morphine and its glucuronides occurs, which accounts for the presence of small amounts of morphine in the feces and in the urine for several days after the last dose. Codeine In contrast to morphine, codeine is approximately 60% as effective orally as parenterally, both as an analgesic and as a respiratory depressant. Codeine, like levorphanol, oxycodone, and methadone, has a high oral to parenteral potency ratio. The greater oral efficacy of these drugs is due to less first-pass metabolism in the liver. Once absorbed, codeine is metabolized by the liver, and its metabolites are excreted chiefly in the urine, largely in inactive forms. A small fraction (approximately 10%) of administered codeine is O-demethylated to form morphine, and both free and conjugated morphine can be found in the urine after therapeutic doses of codeine. Codeine has an exceptionally low affinity for opioid receptors, and the analgesic effect of codeine is due to its conversion to morphine. However, its antitussive actions may involve distinct receptors that bind codeine itself. The half-life of codeine in plasma is 2 to 4 hours. The conversion of codeine to morphine is effected by the cytochrome P450 enzyme CYP2D6. Well-characterized genetic polymorphisms in CYP2D6 lead to the inability to convert codeine to morphine, thus making codeine ineffective as an analgesic for about 10% of the Caucasian population (Eichelbaum and Evert, 1996). Other polymorphisms can lead to enhanced metabolism and thus increased sensitivity to codeine's effects (Eichelbaum and Evert, 1996). Interestingly, there appears to be variation in metabolic efficiency among different ethnic groups. For example, Chinese produce less morphine from codeine than do Caucasians and also are less sensitive to morphine's effects than are Caucasians (Caraco et al., 1999). The reduced sensitivity to morphine may be due to decreased production of morphine-6-glucuronide (Caraco et al., 1999). Thus, it is important to consider the possibility of metabolic enzyme polymorphism in any patient who does not receive adequate analgesia from codeine or an adequate response to other administered prodrugs. Tramadol Tramadol ULTRAM) is a synthetic codeine analog
that is a weak Tramadol is 68% bioavailable after a single oral dose and 100%
available when administered intramuscularly. Its affinity for the Common side effects of tramadol include nausea, vomiting, dizziness, dry mouth, sedation, and headache. Respiratory depression appears to be less than with equianalgesic doses of morphine, and the degree of constipation is less than that seen after equivalent doses of codeine (Duthie, 1998). Tramadol can cause seizures and possibly exacerbate seizures in patients with predisposing factors. While tramadol-induced analgesia is not entirely reversible by naloxone, tramadol-induced respiratory depression can be reversed by naloxone. However, the use of naloxone increases the risk of seizure. Physical dependence on and abuse of tramadol have been reported. Although its abuse potential is unclear, tramadol probably should be avoided in patients with a history of addiction. Because of its inhibitory effect on serotonin uptake, tramadol should not be used in patients taking monoamine oxidase (MAO) inhibitors (Lewis and Han, 1997; see also section on Interactions with Other Drugs, below). Heroin Heroin (diacetylmorphine) is rapidly hydrolyzed to 6-monoacetylmorphine (6-MAM), which, in turn is hydrolyzed to morphine. Both heroin and 6-MAM are more lipid soluble than morphine and enter the brain more readily. Current evidence suggests that morphine and 6-MAM are responsible for the pharmacological actions of heroin. Heroin is mainly excreted in the urine, largely as free and conjugated morphine. The absorption, fate, and distribution of heroin and other morphine-like drugs have been reviewed by (Misra, 1978) and by (Chan and Matzke, 1987). Untoward Effects and Precautions Morphine and related opioids produce a wide spectrum of unwanted effects, including respiratory depression, nausea, vomiting, dizziness, mental clouding, dysphoria, pruritus, constipation, increased pressure in the biliary tract, urinary retention, and hypotension. The bases of these effects have been described above. Rarely, a patient may develop delirium. Increased sensitivity to pain after the analgesia has worn off also may occur. A number of factors may alter a patient's sensitivity to opioid analgesics, including the integrity of the bloodbrain barrier. For example, when morphine is administered to a newborn infant in weight-appropriate doses extrapolated from adults, unexpectedly profound analgesia and respiratory depression may be observed. This is due to the immaturity of the bloodbrain barrier in neonates (Way et al., 1965). As mentioned previously, morphine is hydrophilic, so in the normal situation, proportionately less morphine crosses into the CNS than with more lipophilic opioids. In neonates and in other situations with a compromised bloodbrain barrier, lipophilic opioids may give more predictable clinical results than morphine. In adults, the duration of the analgesia produced by morphine increases progressively with age; however, the degree of analgesia that is obtained with a given dose changes little. Changes in pharmacokinetic parameters only partially explain these observations. The patient with severe pain may tolerate larger doses of morphine. However, as the pain subsides, the patient may exhibit sedation and even respiratory depression as the stimulatory effects of pain are diminished. The reasons for this effect are unclear. All the opioid analgesics are metabolized by the liver, and the drugs should be used with caution in patients with hepatic disease, since increased bioavailability after oral administration or cumulative effects may occur (see Sawe et al., 1981). Renal disease also significantly alters the pharmacokinetics of morphine, codeine, drocode (dihydrocodeine), meperidine, and propoxyphene. Although single doses of morphine are well tolerated, the active metabolite, morphine-6-glucuronide, may accumulate with continued dosing, and symptoms of opioid overdose may result (see Chan and Matzke, 1987). This metabolite also may accumulate during repeated administration of codeine to patients with impaired renal function. When repeated doses of meperidine are given to such patients, the accumulation of normeperidine may cause tremor and seizures (Kaiko et al., 1983). Similarly, the repeated administration of propoxyphene may lead to naloxone-insensitive cardiac toxicity caused by the accumulation of norpropoxyphene (see Chan and Matzke, 1987). Morphine and related opioids must be used cautiously in patients with compromised respiratory function, such as those with emphysema, kyphoscoliosis, or severe obesity. In patients with chronic cor pulmonale, death has occurred following therapeutic doses of morphine. Although many patients with such conditions seem to be functioning within normal limits, they are already utilizing compensatory mechanisms, such as increased respiratory rate. Many have chronically elevated levels of plasma CO2 and may be less sensitive to the stimulating actions of CO2. The further imposition of the depressant effects of opioids can be disastrous. The respiratory-depressant effects of opioids and the related capacity to elevate intracranial pressure must be considered in the presence of head injury or of an already elevated intracranial pressure. While head injury per se does not constitute an absolute contraindication to the use of opioids, the possibility of exaggerated depression of respiration and the potential need to control ventilation of the patient must be considered. Finally, since opioids may produce mental clouding and side effects such as miosis and vomiting, which are important signs in following the clinical course of patients with head injuries, the advisability of their use must be weighed carefully against these risks. Morphine causes histamine release, which can cause bronchoconstriction
and vasodilation. Morphine has the potential to precipitate or exacerbate
asthmatic attacks. The use of morphine should be avoided in patients with a
history of asthma. Other Patients with reduced blood volume are considerably more susceptible to the vasodilatory effects of morphine and related drugs, and these agents must be used cautiously in patients with hypotension from any cause. Allergic phenomena occur with opioid analgesics, but they are not common. They usually are manifested as urticaria and other types of skin rashes such as fixed eruptions; contact dermatitis in nurses and pharmaceutical workers also occurs. Wheals at the site of injection of morphine, codeine, and related drugs are probably secondary to the release of histamine. Anaphylactoid reactions have been reported after intravenous administration of codeine and morphine, but such reactions are rare. It has been suggested, but not proven, that such reactions are responsible for some of the sudden deaths, episodes of pulmonary edema, and other complications that occur among addicts who use heroin intravenously (see Chapter 24: Drug Addiction and Drug Abuse). Interactions with Other Drugs The depressant effects of some opioids may be exaggerated and prolonged by phenothiazines, monoamine oxidase inhibitors, and tricyclic antidepressants; the mechanisms of these supraadditive effects are not fully understood but may involve alterations in the rate of metabolic transformation of the opioid or alterations in neurotransmitters involved in the actions of opioids. Some, but not all, phenothiazines reduce the amount of opioid required to produce a given level of analgesia. However, depending on the specific agent, the respiratory-depressant effects also seem to be enhanced, the degree of sedation is increased, and the hypotensive effects of phenothiazines become an additional complication. Some phenothiazine derivatives enhance the sedative effects, but at the same time seem to be antianalgesic and increase the amount of opioid required to produce satisfactory relief from pain. Small doses of amphetamine substantially increase the analgesic and euphoriant effects of morphine and may decrease its sedative side effects. A number of antihistamines exhibit modest analgesic actions; some (e.g., hydroxyzine) enhance the analgesic effects of low doses of opioids (Rumore and Schlichting, 1986). Antidepressants such as desipramine and amitriptyline are used in the treatment of chronic neuropathic pain but have limited intrinsic analgesic actions in acute pain. However, antidepressants may enhance morphine-induced analgesia (Levine et al., 1986; Pick et al., 1992b). The analgesic synergism between opioids and aspirin-like drugs is discussed below and in Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout. |
Other ![]() -Receptor Agonists
-Receptor Agonists
|
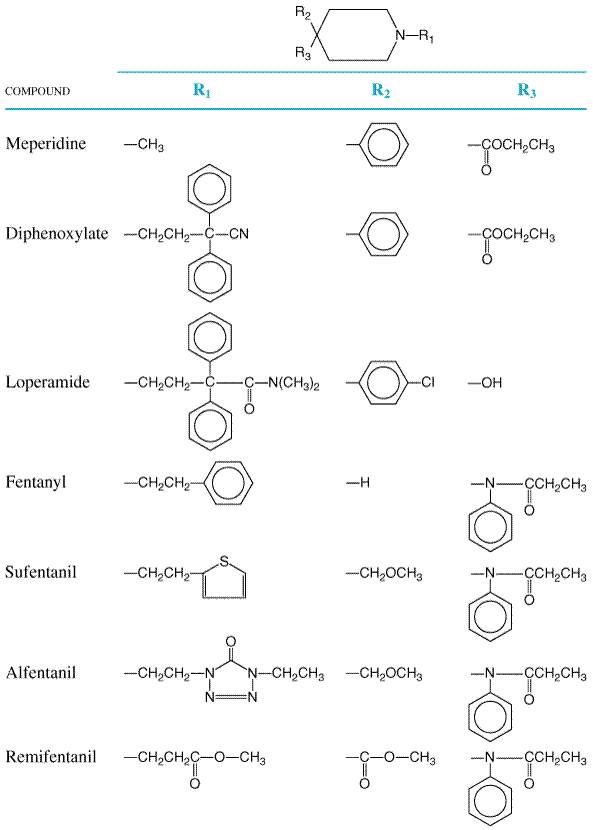
Levorphanol and Congeners Levorphanol (LEVO-DROMORAN) is the only commercially available opioid agonist of the morphinan series. The d-isomer (dextrorphan) is relatively devoid of analgesic action but may have inhibitory effects at NMDA receptors. The structure of levorphanol is shown in Table 235. The pharmacological effects of levorphanol closely parallel those of morphine. However, clinical reports suggest that it may produce less nausea and vomiting. Although levorphanol is less effective when given orally, its oralparenteral potency ratio is comparable to that of codeine and oxycodone. The average adult dose (2 mg subcutaneously) produces analgesia for a period of time somewhat longer than that for morphine. Levorphanol is metabolized less rapidly and has a half-life of about 12 to 16 hours; repeated administration at short intervals may thus lead to accumulation of the drug in plasma (Foley, 1985). Meperidine and Congeners The structural formulas of meperidine, a phenylpiperidine,
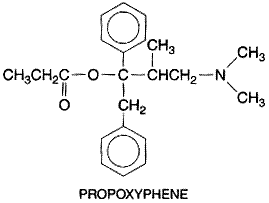
and some of its congeners are shown in Figure 234. Meperidine is
predominantly a
Pharmacological Properties Central Nervous System Meperidine produces a pattern of effects similar but not identical to that described for morphine. Analgesia The analgesic effects of meperidine are detectable about 15 minutes after oral administration, reach a peak in about 1 to 2 hours, and subside gradually. The onset of analgesic effect is faster (within 10 minutes) after subcutaneous or intramuscular administration, and the effect reaches a peak in about 1 hour that corresponds closely to peak concentrations in plasma. In clinical use, the duration of effective analgesia is approximately 1.5 to 3 hours. In general, 75 to 100 mg of meperidine hydrochloride (pethidine, DEMEROL) given parenterally is approximately equivalent to 10 mg of morphine, and, in equianalgesic doses, meperidine produces as much sedation, respiratory depression, and euphoria as does morphine. In terms of total analgesic effect, meperidine is about one-third as effective when given by mouth as when administered parenterally. A few patients may experience dysphoria. Other CNS Actions Peak respiratory depression is observed within 1 hour after intramuscular administration, and there is a return toward normal starting at about 2 hours. Like other opioids, meperidine causes pupillary constriction, increases the sensitivity of the labyrinthine apparatus, and has effects on the secretion of pituitary hormones similar to those of morphine. Meperidine sometimes causes CNS excitation, characterized by tremors, muscle twitches, and seizures; these effects are due largely to accumulation of a metabolite, normeperidine (see below). As with morphine, respiratory depression is responsible for an accumulation of CO2, which, in turn, leads to cerebrovascular dilation, increase in cerebral blood flow, and elevation of cerebrospinal fluid pressure. Cardiovascular System The effects of meperidine on the cardiovascular system generally resemble those of morphine, including the ability to release histamine upon parenteral administration (Lee et al., 1976). Intramuscular administration of meperidine does not significantly affect heart rate, but intravenous administration frequently produces a marked increase in heart rate. Smooth Muscle Meperidine has effects on certain smooth muscles qualitatively similar to those observed with other opioids. Meperidine does not cause as much constipation as does morphine even when given over prolonged periods of time; this may be related to its greater ability to enter the CNS, thereby producing analgesia at lower systemic concentrations. As with other opioids, clinical doses of meperidine slow gastric emptying sufficiently to delay absorption of other drugs significantly. The uterus of a nonpregnant woman usually is mildly stimulated by meperidine. Administered prior to an oxytocic, meperidine does not exert any antagonistic effect. Therapeutic doses given during active labor do not delay the birth process; in fact, the frequency, duration, and amplitude of uterine contraction sometimes may be increased (Zimmer et al., 1988). The drug does not interfere with normal postpartum contraction or involution of the uterus, and it does not increase the incidence of postpartum hemorrhage. Absorption, Fate, and Excretion Meperidine is absorbed by all routes of administration, but the rate of absorption may be erratic after intramuscular injection. The peak plasma concentration usually occurs at about 45 minutes, but the range is wide. After oral administration, only about 50% of the drug escapes first-pass metabolism to enter the circulation, and peak concentrations in plasma are usually observed in 1 to 2 hours (Herman et al., 1985). Meperidine is metabolized chiefly in the liver, with a half-life of about 3 hours. In patients with cirrhosis, the bioavailability of meperidine is increased to as much as 80%, and the half-lives of both meperidine and normeperidine are prolonged. Approximately 60% of meperidine in plasma is protein bound. In human beings, meperidine is hydrolyzed to meperidinic acid, which, in turn, is partially conjugated. Meperidine also is N-demethylated to normeperidine, which may then be hydrolyzed to normeperidinic acid and subsequently conjugated. The clinical significance of the formation of normeperidine is discussed further below. Only a small amount of meperidine is excreted unchanged. Untoward Effects, Precautions, and Contraindications The pattern and overall incidence of untoward effects that follow the use of meperidine are similar to those observed after equianalgesic doses of morphine, except that constipation and urinary retention may be less common. Patients who experience nausea and vomiting with morphine may not do so with meperidine; the converse also may be true. As with other opioids, tolerance develops to some of these effects. The contraindications are generally the same as for other opioids. In patients or addicts who are tolerant to the depressant effects of meperidine, large doses repeated at short intervals may produce an excitatory syndrome including hallucinations, tremors, muscle twitches, dilated pupils, hyperactive reflexes, and convulsions. These excitatory symptoms are due to the accumulation of normeperidine, which has a half-life of 15 to 20 hours compared with 3 hours for meperidine. Opioid antagonists can block the convulsant effect of normeperidine in the mouse. Since normeperidine is eliminated by both the kidney and the liver, decreased renal or hepatic function increases the likelihood of such toxicity (Kaiko et al., 1983). Thus, meperidine is not the drug of choice for the treatment of severe or prolonged pain because of its shorter duration of action relative to morphine and the potential for CNS toxicity from normeperidine. Interaction with Other Drugs Severe reactions may follow the administration of meperidine to patients being treated with MAO inhibitors. Two basic types of interactions can be observed. The most prominent is an excitatory reaction with delirium, hyperthermia, headache, hyper- or hypotension, rigidity, convulsions, coma, and death. This reaction may be due to the ability of meperidine to block neuronal reuptake of serotonin and the resultant serotonergic overactivity (Stack et al., 1988). Therefore, meperidine and its congeners should not be used in patients taking MAO inhibitors. Dextromethorphan also inhibits neuronal serotonin uptake and should be avoided in these patients. As discussed above, tramadol inhibits uptake of norepinephrine and serotonin and should not be used concomitantly with MAO inhibitors. Similar interactions with other currently used opioids have not been observed clinically. Another type of interaction, a potentiation of opioid effect due to inhibition of hepatic microsomal enzymes, also can be observed in patients taking MAO inhibitors, necessitating a reduction in the doses of opioids. Chlorpromazine increases the respiratory-depressant effects of meperidine, as do tricyclic antidepressants; this is not true of diazepam. Concurrent administration of drugs such as promethazine or chlorpromazine also may greatly enhance meperidine-induced sedation without slowing clearance of the drug. Treatment with phenobarbital or phenytoin increases systemic clearance and decreases oral bioavailability of meperidine; this is associated with an elevation of the concentration of normeperidine in plasma (Edwards et al., 1982). As with morphine, concomitant administration of amphetamine has been reported to enhance the analgesic effects of meperidine and its congeners while counteracting sedation. Therapeutic Uses The major use of meperidine is for analgesia. Unlike morphine and its congeners, meperidine is not used for the treatment of cough or diarrhea. Single doses of meperidine also appear to be effective in the treatment of postanesthetic shivering. Meperidine crosses the placental barrier and even in reasonable analgesic doses causes a significant increase in the percentage of babies who show delayed respiration, decreased respiratory minute volume, or decreased oxygen saturation, or who require resuscitation. Both fetal and maternal respiratory depression induced by meperidine can be treated with naloxone. The fraction of drug that is bound to protein is lower in the fetus; concentrations of free drug thus may be considerably higher than in the mother. Nevertheless, meperidine produces less respiratory depression in the newborn than does an equianalgesic dose of morphine or methadone (Fishburne, 1982). Congeners of Meperidine Diphenoxylate Diphenoxylate is a meperidine congener that has a definite constipating effect in human beings. Its only approved use is in the treatment of diarrhea. Although single doses in the therapeutic range (see below) produce little or no morphine-like subjective effects, at high doses (40 to 60 mg) the drug shows typical opioid activity, including euphoria, suppression of morphine abstinence, and a morphine-like physical dependence after chronic administration. Diphenoxylate is unusual in that even its salts are virtually insoluble in aqueous solution, thus obviating the possibility of abuse by the parenteral route. Diphenoxylate hydrochloride is available only in combination with atropine sulfate (LOMOTIL, others). The recommended daily dosage of diphenoxylate for treatment of diarrhea in adults is 20 mg, in divided doses. Difenoxin (difenoxylic acid; MOTOFEN) is one of the metabolites of diphenoxylate; it has actions similar to those of the parent compound. Loperamide Loperamide (IMODIUM, others), like diphenoxylate, is a piperidine derivative (see Figure 233). It slows gastrointestinal motility by effects on the circular and longitudinal muscles of the intestine, presumably as a result of its interactions with opioid receptors in the intestine. Some part of its antidiarrheal effect may be due to a reduction of gastrointestinal secretion (see above; see also Manara and Bianchetti, 1985; Coupar, 1987; Kromer, 1988). In controlling chronic diarrhea, loperamide is as effective as diphenoxylate. In clinical studies, the most common side effect is abdominal cramps. Little tolerance develops to its constipating effect. In human volunteers taking large doses of loperamide, concentrations of drug in plasma peak about 4 hours after ingestion; this long latency may be due to inhibition of gastrointestinal motility and to enterohepatic circulation of the drug. The apparent elimination half-life is 7 to 14 hours. Loperamide is not well absorbed after oral administration and, in addition, apparently does not penetrate well into the brain because of exclusion by a P-glycoprotein transporter widely expressed in the bloodbrain barrier (Sadeque et al., 2000). Mice with deletions of one of the genes encoding the P-glycoprotein transporter have much higher brain levels and significant central effects after administration of loperamide (Schinkel et al., 1996). Inhibition of P-glycoprotein by many clinically used drugs, such as quinidine, verapamil, and ketoconazole, possibly could lead to enhanced central effects of loperamide. In general, loperamide is unlikely to be abused parenterally because of its low solubility; large doses of loperamide given to human volunteers do not elicit pleasurable effects typical of opioids. The usual dosage is 4 to 8 mg per day; the daily dose should not exceed 16 mg. Fentanyl and Congeners Fentanyl
is a synthetic opioid related to the phenylpiperidines (see Figure
233). It is a The actions of fentanyl and its congeners, sufentanil, alfentanil,
and remifentanil, are similar to those of other Pharmacological Properties Analgesia The analgesic effects of fentanyl and sufentanil are similar to those
of morphine and other Other CNS Effects As with other Cardiovascular System Fentanyl and its derivatives decrease the heart rate and can mildly decrease blood pressure. However, these drugs do not release histamine and in general provide a marked degree of cardiovascular stability. Direct depressant effects on the myocardium are minimal. For this reason, high doses of fentanyl or sufentanil commonly are used as the primary anesthetic for patients undergoing cardiovascular surgery or for patients with poor cardiac function. Absorption, Fate, and Excretion These agents are highly lipid soluble and rapidly cross the bloodbrain barrier. This is reflected in the half-life for equilibration between the plasma and CSF of approximately 5 minutes for fentanyl and sufentanil. The levels in plasma and CSF rapidly decline due to redistribution of fentanyl from highly perfused tissue groups to other tissues, such as muscle and fat. As saturation of less-well-perfused tissue occurs, the duration of fentanyl's and sufentanil's effects approach the length of their elimination half lives of between 3 and 4 hours (Sanford and Gutstein, 1995). Fentanyl and sufentanil undergo hepatic metabolism and renal excretion. Therefore, with the use of higher doses or prolonged infusions, fentanyl and sufentanil become longer acting. Therapeutic Uses Fentanyl citrate SUBLIMAZE) and sufentanil citrate (SUFENTA) have gained widespread popularity as anesthetic adjuvants (see Chapter 14: General Anesthetics). They commonly are used either intravenously, epidurally, or intrathecally. A formulation of fentanyl and droperidol (INNOVAR) was commonly used for anesthesia. However, dysphoric side effects of droperidol have limited the popularity of this combination. Epidural use of fentanyl and sufentanil for postoperative or labor analgesia has gained increasing popularity. A combination of epidural opioids with local anesthetics permits reduction in the dosage of both components, minimizing the side effects of both local anesthetics (i.e., motor blockade) and opioids (i.e., urinary retention, itching, and delayed respiratory depression in the case of morphine). Intravenous use of fentanyl and sufentanil for postoperative pain has been effective but limited by clinical concerns about muscle rigidity. However, the use of fentanyl and sufentanil in chronic pain treatment has become more widespread. Epidural and intrathecal infusions, both with and without local anesthetic, are used in the management of chronic malignant pain and selected cases of nonmalignant pain. Also, the development of novel, less invasive routes of administration for fentanyl has facilitated the use of these compounds in chronic pain management. Transdermal patches (DURAGESIC) that provide sustained release of fentanyl for 48 hours or more are available. However, factors promoting increased absorption (e.g., fever) can lead to relative overdosage and increased side effects (see also the section on Alternative Routes of Administration, below). Also, the FENTANYL ORALET, a formulation that permits rapid absorption of fentanyl through the buccal mucosa (much like a lollipop), was tried as an anesthetic premedicant but did not gain wide acceptance due to undesirable side effects in opioid-nave patients (nausea, vomiting, pruritus, and respiratory depression). A similar fentanyl product, ACTIQ, is available in higher strengths and is used for relief of breakthrough cancer pain (Ashburn et al., 1989). Alfentanil and Remifentanil These compounds were developed in an effort to create analgesics with a more rapid onset and predictable termination of opioid effects. The potency of remifentanil is approximately equal to that of fentanyl and is between 20- and 30-times greater than that of alfentanil. The pharmacological properties of alfentanil and remifentanil are similar to those of fentanyl and sufentanil. They have similar incidences of nausea, vomiting, and dose-dependent muscle rigidity. Nausea, vomiting, itching, and headaches have been reported when remifentanil has been used for conscious analgesia for painful procedures. Intracranial pressure changes are minimal when ventilation is controlled. Seizures after remifentanil administration have not yet been reported. Absorption, Fate, and Excretion Both alfentanil and remifentanil have a more rapid onset of analgesic action than do fentanyl on sufentanil. Analgesic effects occur within of 1 to 1.5 minutes. After intravenous administration, alfentanil is metabolized in the liver similarly to fentanyl and sufentanil, with an elimination half-life of 1 to 2 hours. The duration of action of alfentanil is dependent on both the dose and length of administration. Remifentanil is unique in that it is metabolized by plasma esterases (Burkle et al., 1996). Elimination is independent of hepatic metabolism or renal excretion, and the elimination half-life is 8 to 20 minutes. There is no prolongation of effect with repeated dosing or prolonged infusion. Age and weight can affect clearance of remifentanil, requiring that dosage be reduced in the elderly and based on lean body mass. However, neither of these conditions causes major changes in duration of effect. After 3- to 5-hour infusions of remifentanil, recovery of respiratory function can be seen within 3 to 5 minutes, while full recovery from all effects of remifentanil is observed within 15 minutes (Glass et al., 1999). The primary metabolite, remifentanil acid, is 2000- to 4000-times less potent than remifentanil and is renally excreted. Peak respiratory depression after bolus doses of remifentanil occurs after 5 minutes (Patel and Spencer, 1996). Therapeutic Uses Alfentanil hydrochloride ALFENTA) and remifentanil hydrochloride (ULTIVA) are useful for short, painful procedures that require intense analgesia and blunting of stress responses. The titratability of remifentanil and its consistent, rapid offset make it ideally suited for short surgical procedures where rapid recovery is an issue. Remifentanil also has been used successfully for longer neurosurgical procedures, where rapid emergence from anesthesia is important. However, in cases where postprocedural analgesia is required, remifentanil alone is a poor choice. In this situation, either a longer-acting opioid or another analgesic modality should be combined with remifentanil for prolonged analgesia, or another opioid should be used. Alfentanil has been administered intraspinally for pain control. Remifentanil is presently not used intraspinally, as glycine in the drug vehicle can cause temporary motor paralysis. It is generally given by continuous intravenous infusion, as its short duration of action makes bolus administration impractical. Methadone and Congeners Methadone is a long-lasting Chemistry Methadone has the following structural formula:
The analgesic activity of the racemate is almost entirely the result of its content of l-methadone, which is 8- to 50-times more potent than the d isomer; d-methadone also lacks significant respiratory depressant action and addiction liability, but it does possess antitussive activity. Pharmacological Actions The outstanding properties of methadone are its analgesic activity, its efficacy by the oral route, its extended duration of action in suppressing withdrawal symptoms in physically dependent individuals, and its tendency to show persistent effects with repeated administration. Miotic and respiratory-depressant effects can be detected for more than 24 hours after a single dose and, upon repeated administration, marked sedation is seen in some patients. Effects on cough, bowel motility, biliary tone, and the secretion of pituitary hormones are qualitatively similar to those of morphine. Absorption, Fate, and Excretion Methadone is well absorbed from the gastrointestinal tract and can be detected in plasma within 30 minutes after oral ingestion; it reaches peak concentrations at about 4 hours. After therapeutic doses, about 90% of methadone is bound to plasma proteins. Peak concentrations occur in the brain within 1 or 2 hours after subcutaneous or intramuscular administration, and this correlates well with the intensity and duration of analgesia. Methadone also can be absorbed from the buccal mucosa (Weinberg et al., 1988). Methadone undergoes extensive biotransformation in the liver. The major metabolites, the results of N-demethylation and cyclization to form pyrrolidines and pyrroline, are excreted in the urine and the bile along with small amounts of unchanged drug. The amount of methadone excreted in the urine is increased when the urine is acidified. The half-life of methadone is approximately 15 to 40 hours. Methadone appears to be firmly bound to protein in various tissues, including brain. After repeated administration there is gradual accumulation in tissues. When administration is discontinued, low concentrations are maintained in plasma by slow release from extravascular binding sites (see Kreek, 1979); this process probably accounts for the relatively mild but protracted withdrawal syndrome. Side Effects, Toxicity, Drug Interactions, and Precautions Side effects, toxicity, and conditions that alter sensitivity, as well as the treatment of acute intoxication, are similar to those described for morphine. During long-term administration, there may be excessive sweating, lymphocytosis, and increased concentrations of prolactin, albumin, and globulins in the plasma. Rifampin and phenytoin accelerate the metabolism of methadone and can precipitate withdrawal symptoms (see Kreek, 1979). Tolerance and Physical Dependence Volunteer postaddicts who receive subcutaneous or oral methadone daily develop partial tolerance to the nauseant, anorectic, miotic sedative, respiratory-depressant, and cardiovascular effects of methadone. Tolerance develops more slowly to methadone than to morphine in some patients, especially with respect to the depressant effects. However, this may be related in part to cumulative effects of the drug or its metabolites. Tolerance to the constipating effect of methadone does not develop as fully as does tolerance to other effects. The behavior of addicts who use methadone parenterally is strikingly similar to that of morphine addicts, but many former heroin users treated with oral methadone show virtually no overt behavioral effects. Development of physical dependence during the long-term administration of methadone can be demonstrated by drug withdrawal or by administration of an opioid antagonist. Subcutaneous administration of 10 to 20 mg of methadone to former opioid addicts produces definite euphoria equal in duration to that caused by morphine, and its overall abuse potential is comparable to that of morphine. Therapeutic Uses The primary uses of methadone hydrochloride (DOLOPHINE, others) are relief of chronic pain, treatment of opioid abstinence syndromes, and treatment of heroin users. It is not widely used as an antiperistaltic agent. It should not be used in labor. Analgesia The onset of analgesia occurs 10 to 20 minutes following parenteral administration and 30 to 60 minutes after oral medication. The average minimal effective analgesic concentration in blood is about 30 ng/ml (Gourlay et al., 1986). The typical oral dose is 2.5 to 15 mg, depending on the severity of the pain and the response of the patient. The initial parenteral dose is usually 2.5 to 10 mg. Care must be taken when escalating the dosage, because of the prolonged half-life of the drug and its tendency to accumulate over a period of several days with repeated dosing. Despite its longer plasma half-life, the duration of the analgesic action of single doses is essentially the same as that of morphine. With repeated usage, cumulative effects are seen, so that either lower dosage or longer intervals between doses become possible. In contrast to morphine, methadone and many of its congeners retain a considerable degree of their effectiveness when given orally. In terms of total analgesic effects, methadone given orally is about 50% as effective as the same dose administered intramuscularly; however, the oral-parenteral potency ratio is considerably lower when peak analgesic effect is considered. In equianalgesic doses, the pattern and incidence of untoward effects caused by methadone and morphine are similar. Levomethadyl Acetate Levomethadyl acetate (l- Propoxyphene Propoxyphene is structurally related to methadone (see below). Its analgesic effect resides in the dextro isomer, d-propoxyphene (dextropropoxyphene). However, levopropoxyphene seems to have some antitussive activity. The structure of propoxyphene is shown below.
Pharmacological Actions Although slightly less selective than morphine, propoxyphene binds
primarily to As an analgesic, propoxyphene is about one-half to two-thirds as potent as codeine given orally. Ninety to 120 mg of propoxyphene hydrochloride administered orally would equal the analgesic effects of 60 mg of codeine, a dose that usually produces about as much analgesia as 600 mg of aspirin. Combinations of propoxyphene and aspirin, like combinations of codeine and aspirin, afford a higher level of analgesia than does either agent given alone (Beaver, 1988). Absorption, Fate, and Excretion Following oral administration, concentrations of propoxyphene in plasma reach their highest values at 1 to 2 hours. There is great variability between subjects in the rate of clearance and the plasma concentrations that are achieved. The average half-life of propoxyphene in plasma after a single dose is from 6 to 12 hours, which is longer than that of codeine. In human beings, the major route of metabolism is N-demethylation to yield norpropoxyphene. The half-life of norpropoxyphene is about 30 hours, and its accumulation with repeated doses may be responsible for some of the observed toxicity (see Chan and Matzke, 1987). Toxicity Given orally, propoxyphene is approximately one-third as potent as orally administered codeine in depressing respiration. Moderately toxic doses usually produce CNS and respiratory depression, but with still-larger doses the clinical picture may be complicated by convulsions in addition to respiratory depression. Delusions, hallucinations, confusion, cardiotoxicity, and pulmonary edema also have been noted. Respiratory-depressant effects are significantly enhanced when ethanol or sedative-hypnotics are ingested concurrently. Naloxone antagonizes the respiratory-depressant, convulsant, and some of the cardiotoxic effects of propoxyphene. Tolerance and Dependence Very large doses [800 mg of propoxyphene hydrochloride (DARVON, others) or 1200 mg of the napsylate (DARVON-N) per day] reduce the intensity of the morphine withdrawal syndrome somewhat less effectively than do 1500-mg doses of codeine. Maximal tolerated doses are equivalent to daily doses of 20 to 25 mg of morphine, given subcutaneously. The use of higher doses of propoxyphene is prevented by untoward side effects and the occurrence of toxic psychoses. Very large doses produce some respiratory depression in morphine-tolerant addicts, suggesting that cross-tolerance between propoxyphene and morphine is incomplete. Abrupt discontinuation of chronically administered propoxyphene hydrochloride (up to 800 mg per day, given for almost 2 months) results in mild abstinence phenomena, and large oral doses (300 to 600 mg) produce subjective effects that are considered pleasurable by postaddicts. The drug is quite irritating when administered either intravenously or subcutaneously, so that abuse by these routes results in severe damage to veins and soft tissues. Therapeutic Uses Propoxyphene is recommended for the treatment of mild-to-moderate pain. Given acutely, the commonly prescribed combination of 32 mg of propoxyphene with aspirin may not produce more analgesia than aspirin alone, and doses of 65 mg of the hydrochloride or 100 mg of the napsylate are suggested. Propoxyphene is most often given in combination with aspirin or acetaminophen. The wide popularity of propoxyphene in clinical situations in which codeine was once used is largely a result of unrealistic overconcern about the addictive potential of codeine. |
Acute Opioid Toxicity
|
Acute opioid toxicity may result from clinical overdosage, accidental overdosage in addicts, or attempts at suicide. Occasionally, a delayed type of toxicity may occur from the injection of an opioid into chilled skin areas or in patients with low blood pressure and shock. The drug is not fully absorbed, and, therefore, a subsequent dose may be given. When normal circulation is established, an excessive amount may be absorbed suddenly. It is difficult to define the exact amount of any opioid that is toxic or lethal to human beings. Recent experiences with methadone indicate that, in nontolerant individuals, serious toxicity may follow the oral ingestion of 40 to 60 mg. Older literature suggests that, in the case of morphine, a normal, pain-free adult is not likely to die after oral doses of less than 120 mg or to have serious toxicity with less than 30 mg parenterally. Symptoms and Diagnosis The patient who has taken an overdose of an opioid usually is stuporous or, if a large overdose has been taken, may be in a profound coma. The respiratory rate will be very low or the patient may be apneic, and cyanosis may be present. As respiratory exchange decreases, blood pressure, at first likely to be near normal, will fall progressively. If adequate oxygenation is restored early, the blood pressure will improve; if hypoxia persists untreated, there may be capillary damage, and measures to combat shock may be required. The pupils will be symmetrical and pinpoint in size; however, if hypoxia is severe, they may be dilated. Urine formation is depressed. Body temperature falls, and the skin becomes cold and clammy. The skeletal muscles are flaccid, the jaw is relaxed, and the tongue may fall back and block the airway. Frank convulsions occasionally may be noted in infants and children. When death occurs, it is nearly always due to respiratory failure. Even if respiration is restored, death still may occur as a result of complications that develop during the period of coma, such as pneumonia or shock. Noncardiogenic pulmonary edema is seen commonly with opioid poisoning. It probably is not due to contaminants or to anaphylactoid reactions, and it has been observed following toxic doses of morphine, methadone, propoxyphene, and uncontaminated heroin. The triad of coma, pinpoint pupils, and depressed respiration strongly suggests opioid poisoning. The finding of needle marks suggestive of addiction further supports the diagnosis. Mixed poisonings, however, are not uncommon. Examination of the urine and gastric contents for drugs may aid in diagnosis, but the results usually become available too late to influence treatment. Treatment The first step is to establish a patent airway and ventilate the patient. Opioid antagonists (see Opioid Antagonists) can produce dramatic reversal of the severe respiratory depression, and the antagonist naloxone (see below) is the treatment of choice. However, care should be taken to avoid precipitating withdrawal in dependent patients, who may be extremely sensitive to antagonists. The safest approach is to dilute the standard naloxone dose (0.4 mg) and slowly administer it intravenously, monitoring arousal and respiratory function. With care, it usually is possible to reverse the respiratory depression without precipitating a major withdrawal syndrome. If no response is seen with the first dose, additional doses can be given. Patients should be observed for rebound increases in sympathetic nervous system activity, which may result in cardiac arrhythmias and pulmonary edema (see Duthie and Nimmo, 1987). For reversing opioid poisoning in children, the initial dose of naloxone is 0.01 mg/kg. If no effect is seen after a total dose of 10 mg, one can reasonably question the accuracy of the diagnosis. Pulmonary edema sometimes associated with opioid overdosage may be countered by positive-pressure respiration. Tonic-clonic seizures, occasionally seen as part of the toxic syndrome with meperidine and propoxyphene, are ameliorated by treatment with naloxone. The presence of general CNS depressants does not prevent the salutary effect of naloxone, and in cases of mixed intoxications, the situation will be improved largely due to antagonism of the respiratory-depressant effects of the opioid. However, some evidence indicates that naloxone and naltrexone also may antagonize some of the depressant actions of sedative-hypnotics. One need not attempt to restore the patient to full consciousness. The duration of action of the available antagonists is shorter than that of many opioids; hence, patients must be watched carefully, lest they slip back into coma. This is particularly important when the overdosage is due to methadone or l-acetylmethadol. The depressant effects of these drugs may persist for 24 to 72 hours, and fatalities have occurred as a result of premature discontinuation of naloxone. In cases of overdoses of these drugs, a continuous infusion of naloxone should be considered. Toxicity due to overdose of pentazocine and other opioids with mixed actions may require higher doses of naloxone. The pharmacological actions of opioid antagonists are discussed in more detail in Opioid Antagonists. |
Opioid Agonist/Antagonists and Partial Agonists
|
The drugs described in this section differ
from clinically used Pentazocine Pentazocine was synthesized as part of a deliberate effort to develop an effective analgesic with little or no abuse potential. It has both agonistic actions and weak opioid antagonistic activity. The pharmacology of pentazocine has been reviewed by (Brogden et al., 1973). Chemistry Pentazocine is a benzomorphan derivative with the following structural formula:
The compound has a large substituent on the nitrogen atom that is analogous to position 17 of morphine. This structural feature is common to a number of opioids with antagonist or agonist/ antagonist activity. The analgesic and respiratory-depressant activity of the racemate is due mainly to the l isomer. Pharmacological Actions The pattern of CNS effects produced by pentazocine is generally
similar to that of the morphine-like opioids, including analgesia, sedation,
and respiratory depression. The analgesic effects of pentazocine are due to agonistic
actions at The cardiovascular responses to pentazocine differ from those seen
with typical Pentazocine acts as a weak antagonist or partial agonist at Absorption, Fate, and Excretion Pentazocine is well absorbed from the gastrointestinal tract and from subcutaneous and intramuscular sites. Peak analgesia occurs 15 minutes to 1 hour after intramuscular administration and 1 to 3 hours after oral administration. The half-life in plasma is 4 to 5 hours. First-pass metabolism in the liver is extensive, and somewhat less than 20% of pentazocine enters the systemic circulation. Drug action is terminated by hepatic metabolism and renal excretion. Side Effects, Toxicity, and Precautions The most commonly reported untoward effects are sedation, sweating, and dizziness or lightheadedness; nausea also occurs, but vomiting is less common than with morphine. Psychotomimetic effects, such as uncontrollable or weird thoughts, anxiety, nightmares, and hallucinations, occur with parenteral doses above 60 mg. Epidemiological data suggest that overdose with pentazocine alone rarely causes death. High doses produce marked respiratory depression associated with increased blood pressure and tachycardia. The respiratory depression is antagonized by naloxone. Pentazocine is irritating when administered subcutaneously or intramuscularly. Repeated injections over long periods may cause extensive fibrosis of subcutaneous and muscular tissue. Patients who have been receiving opioids on a regular basis may experience abstinence signs and symptoms when given pentazocine. After an opioid-free interval of 1 to 2 days, it is usually possible to administer pentazocine without producing such withdrawal effects. Tolerance and Physical Dependence With frequent and repeated use, tolerance develops to the analgesic
and subjective effects of pentazocine. However, pentazocine does not prevent
or ameliorate the morphine withdrawal syndrome. Instead, when high doses of
pentazocine are given to subjects dependent on morphine, it precipitates
withdrawal symptoms because of its antagonistic actions at the After long-term administration (60 mg every 4 hours), postaddicts
develop physical dependence that can be demonstrated by abrupt withdrawal or
by the administration of naloxone. The withdrawal syndrome after chronic
doses of more than 500 mg per day, although milder in intensity than
withdrawal from morphine, includes abdominal cramps, anxiety, chills,
elevated temperature, vomiting, lacrimation, and sweating. Pentazocine
withdrawal symptoms can be managed by gradual reduction of pentazocine itself
or by substitution of Therapeutic Uses Pentazocine is used as an analgesic. Although the risk of drug dependence exists, it may be lower than that associated with the use of morphine-like drugs in similar circumstances. Because abuse patterns appear to be less likely to develop with oral administration, this route should be used whenever possible. Pentazocine lactate TALWIN) is available as a solution for injection. In an effort to reduce the use of tablets as a source of injectable pentazocine, tablets for oral use now contain pentazocine hydrochloride (equivalent to 50 mg of the base) and naloxone hydrochloride (equivalent to 0.5 mg of the base; TALWIN NX). After oral ingestion, naloxone is destroyed rapidly by the liver; however, if the material is dissolved and injected, the naloxone produces aversive effects in subjects dependent on opioids. Tablets containing mixtures of pentazocine with aspirin (TALWIN COMPOUND) or acetaminophen (TALCEN) also are available. In terms of analgesic effect, 30 to 60 mg of pentazocine given parenterally is approximately equivalent to 10 mg of morphine. An oral dose of about 50 mg of pentazocine results in analgesia equivalent to that produced by 60 mg of codeine orally. Nalbuphine Nalbuphine is related structurally to both naloxone and oxymorphone (see
Table 235). It is an agonist/antagonist opioid with a spectrum of effects
that qualitatively resembles that of pentazocine; however, nalbuphine is a
more potent antagonist at Pharmacological Actions and Side Effects An intramuscular dose of 10 mg of nalbuphine is equianalgesic to 10 mg of morphine, with similar onset and duration of both analgesic and subjective effects. Nalbuphine depresses respiration as much as do equianalgesic doses of morphine. However, nalbuphine exhibits a ceiling effect, such that increases in dosage beyond 30 mg produce no further respiratory depression. However, a ceiling effect for analgesia also is reached at this point. In contrast to pentazocine and butorphanol, 10 mg of nalbuphine given to patients with stable coronary artery disease does not produce an increase in cardiac index, pulmonary arterial pressure, or cardiac work, and systemic blood pressure is not significantly altered; these indices also are relatively stable when nalbuphine is given to patients with acute myocardial infarction (see Roth et al., 1988). Its gastrointestinal effects are probably similar to those of pentazocine. Nalbuphine produces few side effects at doses of 10 mg or less; sedation, sweating, and headache are the most common. At much higher doses (70 mg), psychotomimetic side effects (dysphoria, racing thoughts, and distortions of body image) can occur. Nalbuphine is metabolized in the liver and has a half-life in plasma of 2 to 3 hours. Given orally, nalbuphine is 20% to 25% as potent as when given intramuscularly. Tolerance and Physical Dependence In subjects dependent on low doses of morphine (60 mg per day), nalbuphine
precipitates an abstinence syndrome. Prolonged administration of nalbuphine
can produce physical dependence. The withdrawal syndrome is similar in
intensity to that seen with pentazocine. The potential for abuse of
parenteral nalbuphine in subjects not dependent on Therapeutic Uses Nalbuphine hydrochloride NUBAIN) is used to produce analgesia. Because it is an agonist/antagonist, administration to patients who have been receiving morphine-like opioids may create difficulties unless a brief drug-free interval is interposed. The usual adult dose is 10 mg parenterally every 3 to 6 hours; this may be increased to 20 mg in nontolerant individuals. Butorphanol Butorphanol is a morphinan congener with a profile of actions similar to those of pentazocine. The structural formula of butorphanol is shown in Table 235. Pharmacological Actions and Side Effects In postoperative patients, a parenteral dose of 2 to 3 mg of butorphanol produces analgesia and respiratory depression approximately equal to that produced by 10 mg of morphine or 80 to 100 mg of meperidine; the onset, peak, and duration of action are similar to those that follow the administration of morphine. The plasma half-life of butorphanol is about 3 hours. Like pentazocine, analgesic doses of butorphanol produce an increase in pulmonary arterial pressure and in the work of the heart; systemic arterial pressure is slightly decreased (Popio et al., 1978). The major side effects of butorphanol are drowsiness, weakness, sweating, feelings of floating, and nausea. While the incidence of psychotomimetic side effects is lower than that with equianalgesic doses of pentazocine, they are qualitatively similar. Physical dependence on butorphanol can occur. Therapeutic Uses Butorphanol tartrate STADOL) is
better suited for the relief of acute rather than chronic pain. Because of
its side effects on the heart, it is less useful than morphine or meperidine
in patients with congestive heart failure or myocardial infarction. The usual
dose is between 1 and 4 mg of the tartrate given intramuscularly, or 0.5 to 2
mg given intravenously every 3 to 4 hours. A nasal formulation ( Buprenorphine Buprenorphine is a semisynthetic, highly lipophilic opioid derived from thebaine (see Table 235). It is 25 to 50 times more potent than morphine. Pharmacological Actions and Side Effects Buprenorphine produces analgesia and other CNS effects that are qualitatively similar to those of morphine. About 0.4 mg of buprenorphine is equianalgesic with 10 mg of morphine given intramuscularly (Wallenstein et al., 1986). Although variable, the duration of analgesia is usually longer than that of morphine. Some of the subjective and respiratory-depressant effects are unequivocally slower in onset and longer lasting than those of morphine. For example, peak miosis occurs about 6 hours after intramuscular injection, while maximal respiratory depression is observed at about 3 hours. Buprenorphine appears to be a partial Buprenorphine is relatively well absorbed by most routes. Administered sublingually, the drug (0.4 to 0.8 mg) produces satisfactory analgesia in postoperative patients. Concentrations in blood peak within 5 minutes after intramuscular injection and within 1 to 2 hours after oral or sublingual administration. While the half-life in plasma has been reported to be about 3 hours, this value bears little relationship to the rate of disappearance of effects (see above). Both N-dealkylated and conjugated metabolites are detected in the urine, but most of the drug is excreted unchanged in the feces. About 96% of the circulating drug is bound to protein. Physical Dependence When buprenorphine is discontinued, a withdrawal syndrome develops that is delayed in onset for 2 days to 2 weeks; this consists of typical, but generally not very severe, morphine-like withdrawal signs and symptoms, and it persists for about 1 to 2 weeks (Bickel et al., 1988; Fudala et al., 1989). Therapeutic Uses Buprenorphine. BUPRENEX) may be used as an analgesic and also has proven to be useful as a maintenance drug for opioid-dependent subjects (Johnson et al., 2000). The drug was approved provisionally for use in the treatment of heroin addiction when the Drug Addiction Treatment Act was passed by the United States Congress and signed by the President in October of 2000. Approval by the Food and Drug Administration is pending. The usual intramuscular or intravenous dose for analgesia is 0.3 mg, given every 6 hours. Sublingual doses of 0.4 to 0.8 mg produce effective analgesia, and doses of 6 to 8 mg appear to be about equal to 60 mg of methadone as a maintenance agent. Other Agonist/Antagonists Meptazinol
is an agonist/antagonist opioid that is about one-tenth as potent as morphine
in producing analgesia. Its duration of action is somewhat shorter than that
of morphine. Meptazinol also has cholinergic actions that may contribute to
its analgesic effects (see Holmes and Ward, 1985). Nevertheless, its
analgesic actions are antagonized by naloxone, and it can precipitate
withdrawal in animals dependent on |
Opioid Antagonists
|
Under ordinary circumstances, the drugs to be discussed in this section produce few effects unless opioids with agonistic actions have been administered previously. However, when the endogenous opioid systems are activated, as in shock or certain forms of stress, the administration of an opioid antagonist alone may have visible consequences. These agents have obvious therapeutic utility in the treatment of overdosage with opioids. As the understanding of the role of endogenous opioid systems in pathophysiological states increases, additional therapeutic indications for these antagonists may develop. Chemistry Relatively minor changes in the structure of an opioid can convert a
drug that is primarily an agonist into one with antagonistic actions at one
or more types of opioid receptors. The most common such substitution is that
of a larger moiety (e.g., an allyl or methylcyclopropyl group) for the
N-methyl group that is typical of the Nalmefene REVIX) is a relatively pure Pharmacological Properties If endogenous opioid systems have not been activated, the pharmacological actions of opioid antagonists depend on whether or not an opioid agonist has been administered previously, on the pharmacological profile of that opioid, and on the degree to which physical dependence on an opioid has developed. Effects in the Absence of Opioid Drugs Subcutaneous doses of naloxone (NARCAN; up to 12 mg) produce no discernible subjective effects in human beings, and 24 mg causes only slight drowsiness. Naltrexone (REVIA) also appears to be a relatively pure antagonist but with higher oral efficacy and a longer duration of action. At high doses, both naloxone and naltrexone may have some special agonistic effects. However, these are of little clinical significance. At doses in excess of 0.3 mg/kg of naloxone, normal subjects show increased systolic blood pressure and decreased performance on tests of memory. High doses of naltrexone appeared to cause mild dysphoria in one study but almost no subjective effect in several others (see Gonzalez and Brogden, 1988). Although high doses of antagonists might be expected to alter the
actions of endogenous opioid peptides, the detectable effects are usually
both subtle and limited (Cannon and Liebeskind, 1987). Most likely, this
reflects the low levels of tonic activity of the opioid systems. In this
regard, analgesic effects can be differentiated from endocrine effects, in
which naloxone causes readily demonstrable changes in hormone levels (see
below). It is interesting that naloxone appears to block the analgesic
effects of placebo medications and acupuncture. In laboratory animals, the
administration of naloxone will reverse or attenuate the hypotension
associated with shock of diverse origins including that caused by
anaphylaxis, endotoxin, hypovolemia, and injury to the spinal cord; opioid
agonists aggravate these conditions (Amir, 1988). Naloxone apparently acts to
antagonize the actions of endogenous opioids that are mobilized by pain or
stress and that are involved in the regulation of blood pressure by the CNS.
Although neural damage that follows trauma to the spinal cord or cerebral
ischemia also appears to involve endogenous opioids, it is not certain
whether opioid antagonists can prevent damage to these or other organs and/or
increase rates of survival. Nevertheless, opioid antagonists can reduce the
extent of injury in some animal models, perhaps by blocking As noted above, endogenous opioid peptides participate in the regulation of pituitary secretion, apparently by exerting tonic inhibitory effects on the release of certain hypothalamic hormones (see Chapter 56: Pituitary Hormones and Their Hypothalamic Releasing Factors). Thus, the administration of naloxone or naltrexone increases the secretion of gonadotropin-releasing hormone and corticotropin-releasing factor and elevates the plasma concentrations of LH, FSH, and ACTH, as well as the hormones produced by their target organs. Antagonists do not consistently alter basal or stress-induced concentrations of prolactin in plasma in men; paradoxically, naloxone stimulates the release of prolactin in women. Opioid antagonists augment the increases in plasma concentrations of cortisol and catecholamines that normally accompany stress or exercise. The neuroendocrine effects of opioid antagonists have been reviewed (Howlett and Rees, 1986). Endogenous opioid peptides probably have some role in the regulation of feeding or energy metabolism, because opioid antagonists increase energy expenditure and interrupt hibernation in appropriate species and induce weight loss in genetically obese rats. The antagonists also prevent stress-induced overeating and obesity in rats. These observations have led to the experimental use of opioid antagonists in the treatment of human obesity, especially that associated with stress-induced eating disorders. However, naltrexone does not accelerate weight loss in very obese subjects, even though short-term administration of opioid antagonists reduce food intake in both lean and obese individuals (Atkinson, 1987). Antagonistic Actions Small doses (0.4 to 0.8 mg) of naloxone given intramuscularly or
intravenously prevent or promptly reverse the effects of Effects in Physical Dependence In subjects who are dependent on morphine-like opioids, small
subcutaneous doses of naloxone (0.5 mg) precipitate a moderate-to-severe
withdrawal syndrome that is very similar to that seen after abrupt withdrawal
of opioids, except that the syndrome appears within minutes after
administration and subsides in about 2 hours. The severity and duration of
the syndrome are related to the dose of the antagonist and to the degree and
type of dependence. Higher doses of naloxone will precipitate a withdrawal
syndrome in patients dependent on pentazocine, butorphanol, or nalbuphine.
Naloxone produces 'overshoot' phenomena suggestive of early acute
physical dependence 6 to 24 hours after a single dose of a Tolerance and Physical Dependence Even after prolonged administration of high doses, discontinuation of naloxone is not followed by any recognizable withdrawal syndrome, and the withdrawal of naltrexone, another relatively pure antagonist, produces very few signs and symptoms. However, long-term administration of antagonists increases the density of opioid receptors in the brain and causes a temporary exaggeration of responses to the subsequent administration of opioid agonists (Yoburn et al., 1988). Naltrexone and naloxone have little or no potential for abuse. Absorption, Fate, and Excretion Although absorbed readily from the gastrointestinal tract, naloxone is almost completely metabolized by the liver before reaching the systemic circulation and thus must be administered parenterally. The drug is absorbed rapidly from parenteral sites of injection and is metabolized in the liver, primarily by conjugation with glucuronic acid; other metabolites are produced in small amounts. The half-life of naloxone is about 1 hour, but its clinically effective duration of action can be even less. Compared with naloxone, naltrexone retains much more of its efficacy by the oral route, and its duration of action approaches 24 hours after moderate oral doses. Peak concentrations in plasma are reached within 1 to 2 hours and then decline with an apparent half-life of approximately 3 hours; this value does not change with long-term use. Naltrexone is metabolized to 6-naltrexol, which is a weaker antagonist but has a longer half-life of about 13 hours. Naltrexone is much more potent than naloxone, and l00-mg oral doses given to patients addicted to opioids produce concentrations in tissues sufficient to block the euphorigenic effects of 25-mg intravenous doses of heroin for 48 hours (see Gonzalez and Brogden, 1988). Therapeutic Uses Opioid antagonists have established uses in the treatment of opioid-induced toxicity, especially respiratory depression; in the diagnosis of physical dependence on opioids; and as therapeutic agents in the treatment of compulsive users of opioids, as discussed in Chapter 24: Drug Addiction and Drug Abuse. Their potential utility in the treatment of shock, stroke, spinal cord and brain trauma, and other disorders that may involve mobilization of endogenous opioid peptides remains to be established. Naltrexone is approved by the United States Food and Drug Administration for treatment of alcoholism (see Chapters 18: Ethanol and 24: Drug Addiction and Drug Abuse). Treatment of Opioid Overdosage Naloxone hydrochloride is used to treat opioid overdose. As discussed earlier, it acts
rapidly to reverse the respiratory depression associated with high doses of
opioids. However, it should be used cautiously, since it also can precipitate
withdrawal in dependent subjects and cause undesirable cardiovascular side
effects. By carefully titrating the dose of naloxone, it usually is possible
to antagonize the respiratory-depressant actions without eliciting a full
withdrawal syndrome. The duration of action of naloxone is relatively short,
and it often must be given repeatedly or by continuous infusion. Opioid
antagonists also have been effectively employed to decrease neonatal
respiratory depression secondary to the intravenous or intramuscular
administration of opioids to the mother. In the neonate, the initial dose is
10 |
Centrally Active Antitussive Agents
|
Cough is a useful physiological mechanism that serves to clear the respiratory passages of foreign material and excess secretions. It should not be suppressed indiscriminately. There are, however, many situations in which cough does not serve any useful purpose but may, instead, only annoy the patient or prevent rest and sleep. Chronic cough can contribute to fatigue, especially in elderly patients. In such situations the physician should use a drug that will reduce the frequency or intensity of the coughing. The cough reflex is complex, involving the central and peripheral nervous systems as well as the smooth muscle of the bronchial tree. It has been suggested that irritation of the bronchial mucosa causes bronchoconstriction, which, in turn, stimulates cough receptors (which probably represent a specialized type of stretch receptor) located in tracheobronchial passages. Afferent conduction from these receptors is via fibers in the vagus nerve; central components of the reflex probably involve several mechanisms or centers that are distinct from the mechanisms involved in the regulation of respiration. The drugs that directly or indirectly can affect this complex
mechanism are diverse. For example, cough may be the first or only symptom in
bronchial asthma or allergy, and in such cases bronchodilators (e.g., A number of drugs are known to reduce cough as a result of their central actions, although the exact mechanisms are still not entirely clear. Included among them are the opioid analgesics discussed above (codeine and hydrocodone are the opioids most commonly used to suppress cough), as well as a number of nonopioid agents. Cough suppression often occurs with lower doses of opioids than those needed for analgesia. A 10- or 20-mg oral dose of codeine, although ineffective for analgesia, produces a demonstrable antitussive effect, and higher doses produce even more suppression of chronic cough. In selecting a specific centrally active agent for a particular patient, the significant considerations are its antitussive efficacy against pathological cough and the incidence and type of side effects to be expected. In the majority of situations requiring a cough suppressant, liability for abuse need not be a major consideration. Most of the nonopioid agents now offered as antitussives are effective against cough induced by a variety of experimental techniques. However, the ability of these tests to predict clinical efficacy is limited. Dextromethorphan Dextromethorphan (d-3-methoxy- N-methylmorphinan) is the d isomer of the codeine analog methorphan; however, unlike the l isomer, it has no analgesic or addictive properties and does not act through opioid receptors. The drug acts centrally to elevate the threshold for coughing. Its effectiveness in patients with pathological cough has been demonstrated in controlled studies; its potency is nearly equal to that of codeine. Compared with codeine, dextromethorphan produces fewer subjective and gastrointestinal side effects (Matthys et al., 1983). In therapeutic dosages, the drug does not inhibit ciliary activity, and its antitussive effects persist for 5 to 6 hours. Its toxicity is low, but extremely high doses may produce CNS depression. Sites that bind dextromethorphan with high affinity have been identified in membranes from various regions of the brain (Craviso and Musacchio, 1983). Although dextromethorphan is known to function as an NMDA-receptor antagonist, these binding sites are not limited to the known distribution of NMDA receptors (Elliott et al., 1994). Thus, the mechanism by which dextromethorphan exerts its antitussive effects is still unclear. Two other known antitussives, carbetapentane and caramiphen, also bind avidly to this site, but codeine, levopropoxyphene, and other antitussive opioids (as well as naloxone) are not bound. Although noscapine (see below) enhances the affinity of dextromethorphan, it appears to interact with distinct binding sites (Karlsson et al., 1988). The relationship of these binding sites to antitussive actions is not known; however, these observations, coupled with the ability of naloxone to antagonize the antitussive effects of codeine but not those of dextromethorphan, indicate that cough suppression can be achieved by a number of different mechanisms. The average adult dosage of dextromethorphan hydrobromide is 10 to 30 mg three to six times daily; however, as is the case with codeine, higher doses often are required. The drug is generally marketed for 'over-the-counter' sale in numerous syrups and lozenges or in combinations with antihistamines and other agents. Other Drugs Levopropoxyphene napsylate, the l-isomer of dextropropoxyphene, in doses of 50 to 100 mg orally, appears to suppress cough to about the same degree as does 30 mg of dextromethorphan. Unlike dextropropoxyphene, levopropoxyphene has little or no analgesic activity. Noscapine is a naturally occurring opium alkaloid of the benzylisoquinoline group; except for its antitussive effect, it has no significant actions on the CNS in doses within the therapeutic range. The drug is a potent releaser of histamine, and large doses cause bronchoconstriction and transient hypotension. Other drugs that have been used as centrally acting antitussives include carbetapentane, caramiphen, chlophedianol, diphenhydramine, and glaucine. Each is a member of a distinct pharmacological class unrelated to the opioids. The mechanism of action of diphenhydramine, an antihistamine, is unclear. Although sedative effects are common, paradoxical excitement may be seen in infants; dryness of mucous membranes caused by anticholinergic effects and thickening of mucus may be a disadvantage. In general, the toxicity of these agents is low, but controlled clinical studies are still insufficient to determine whether or not they merit consideration as alternatives to more thoroughly studied agents. Pholcodine
[3-O-(2-morpholinoethyl)morphine] is used clinically in many countries
outside the Benzonatate TESSALON) is a long-chain polyglycol derivative chemically related to procaine and believed to exert its antitussive action on stretch or cough receptors in the lung, as well as by a central mechanism. It has been administered by all routes; the oral dosage is 100 mg three times daily, but higher doses have been used. |
Therapeutic Uses of Opioid Analgesics
|
Sir William Osler called morphine 'God's own medicine.' Opioids are still the mainstay of pain treatment. However, the development of new analgesic compounds and new routes of administration have increased the therapeutic options available to clinicians, while at the same time helping to minimize undesirable side effects. In this section, we will outline guidelines for rational drug selection, discuss routes of administration other than the standard oral and parenteral methods, and outline general principles for the use of opioids in acute and chronic pain states. Extensive efforts by many individuals and organizations have resulted in the publication of many useful guidelines for the administration of opioids. These have been developed for a number of clinical situations, including acute pain, trauma, cancer, nonmalignant chronic pain, and treatment of pain in children (Agency for Health Care Policy and Research, 1992a, 1992b, 1994; International Association for the Study of Pain, 1992; American Pain Society, 1999; Grossman et al., 1999; World Health Organization, 1998; Berde et al., 1990). These guidelines provide comprehensive discussions of dosing regimens and drug selection and also provide protocols for the management of complex conditions. In the case of cancer pain, adherence to standardized protocols for cancer pain management (Agency for Health Care Policy and Research, 1994) has been shown to improve pain management significantly (Du Pen et al., 1999). Guidelines for the oral and parenteral dosing of commonly used opioids are presented in Table 236. These guidelines are for acute pain management in opioid-nave patients. Adjustments will need to be made for use in opioid-tolerant patients and in chronic pain states. For children under 6 months of age, especially those who are ill or premature, expert consultation should be obtained. The pharmacokinetics and potency of opioids can be substantially altered in these patients, and in some cases there is a significant risk of apnea. It also should be noted that there is substantial individual variability in responses to opioids. A standard intramuscular dose of 10 mg of morphine sulfate will relieve severe pain adequately in only 2 of 3 patients. Adjustments will have to be made based on clinical response. In general, it is recommended that opioids always be combined with other analgesic agents, such as nonsteroidal anti-inflammatory drugs (NSAIDS) or acetaminophen. In this way, one can take advantage of additive analgesic effects and minimize the dose of opioids and thus undesirable side effects. In some situations, NSAIDS can provide analgesia equal to that produced by 60 mg of codeine. Potentiation of opioid action by NSAIDs may be due to increased conversion of arachidonic acid to 12-lipoxygenase products that facilitate effects of opioids on K+ channels (Vaughan et al., 1997). This 'opioid-sparing' strategy is the backbone of the 'analgesic ladder' for pain management proposed by the World Health Organization (1990). Weaker opioids can be supplanted by stronger opioids in cases of moderate and severe pain. In addition, analgesics always should be dosed in a continuous or 'around the clock' fashion rather than on an as needed basis for chronic severe pain. This provides more consistent analgesic levels and avoids unnecessary suffering. Knowledge of the pharmacological profiles of analgesics allows the rational selection of dosing intervals without risk of overdosage. Factors guiding the selection of specific opioid compounds for pain treatment include potency, pharmacokinetic characteristics, and the routes of administration available. A more potent compound could be useful when high doses of opioid are required, so the medicine can be given in a smaller volume. Duration of action also is an important consideration. For example, a long-acting opioid such as methadone may be appropriate when less-frequent dosing is desired. For short, painful procedures, a quick-acting, fast-dissipating compound such as remifentanil would be a useful choice. In special cases, where a lower addiction risk is required or in patients unable to tolerate other opioids, a partial agonist or mixed agonist/antagonist compound might be a rational choice. The properties of some commonly used orally-administered opioids are discussed in more detail below. Morphine is available for oral use in standard and controlled-release preparations. Due to first-pass metabolism, morphine is two- to sixfold less potent orally than parenterally. This is important to remember when converting a patient from parenteral to oral medication. There is wide variability in the first-pass metabolism, and the dose should be titrated to the patient's needs. In children who weigh less than 50 kg, morphine can be given at 0.1 mg/kg every 3 to 4 hours parenterally or at 0.3 mg/kg orally. Codeine is widely used only due to its high oral/parenteral potency ratio. Orally, codeine at 30 mg is approximately equianalgesic to 325 to 600 mg of aspirin. Combinations of codeine with aspirin or acetaminophen usually provide additive actions, and at these doses analgesic efficacy can exceed that of 60 mg of codeine (see Beaver, 1988). Many drugs can be used instead of either morphine or codeine, as shown in Table 236. Oxycodone, with its high oral/parenteral potency ratio, is widely used in combination with aspirin (PERCODAN, others) or acetaminophen (PERCOCET 2.5/325, others), although it is available alone (ROXICODINE, others). Heroin (diacetylmorphine) is not available for therapeutic use in the Alternative Routes of Administration In addition to the traditional oral and parenteral formulations for opioids, many other methods have been developed in an effort to improve therapeutic efficacy while minimizing side effects. These routes also improve the ease of use of opioids, and increase patient satisfaction. Patient-Controlled Analgesia (PCA) With this modality, the patient has limited control of the dosing of opioid from an infusion pump within tightly mandated parameters. PCA can be used for intravenous or epidural infusion. This technique avoids any delays in administration and permits greater dosing flexibility than other regimens, better adapting to individual differences in responsiveness to pain and to opioids. It also gives the patient a greater sense of control. With shorter-acting opioids, serious toxicity or excessive use rarely occurs. An early concern that self-administration of opioids would increase the probability of addiction has not materialized. PCA is suitable for both adults and children, and it is preferred over intramuscular injections for postoperative pain control (Rodgers et al., 1988). Computer-Assisted Continuous Infusion (CACI) The idea behind this mode of administration is to enable clinicians to titrate intravenous agents in a fashion similar to that used in delivering volatile agents (Sanford and Gutstein, 1995). CACI based on detailed pharmacokinetic models has been used successfully to administer opioids (Shafer et al., 1990; Bailey et al., 1993). However, true 'closed-loop' control of opioid administration requires the capability of continuously measuring plasma opioid levels with indwelling sensors. Until such real-time measurement is available, accurate assessment of dose-effect relationships in patients is not possible. Intraspinal Infusion Administration of opioids into the epidural or intrathecal space provides more direct access to the first pain-processing synapse in the dorsal horn of the spinal cord. This permits the use of doses substantially lower than those required for oral or parenteral administration (see Table 237). Systemic side effects are thus decreased. However, epidural opioids have their own dose-dependent side effects, such as itching, nausea, vomiting, respiratory depression, and urinary retention. The use of hydrophilic opioids such as preservative-free morphine (DURAMORPH, others) permits more rostral spread of the compound, allowing it to directly affect supraspinal sites. As a consequence, after intraspinal morphine, delayed respiratory depression can be observed for as long as 24 hours after a bolus dose. While the risk of delayed respiratory depression is reduced with more lipophilic opioids, it is not eliminated. Extreme vigilance and appropriate monitoring are required for all patients receiving intraspinal narcotics. Nausea and vomiting also are more prominent symptoms with intraspinal morphine. However, supraspinal analgesic centers also can be stimulated, possibly leading to synergistic analgesic effects. Analogous to the relationship between systemic opioids and NSAIDS, intraspinal narcotics often are combined with local anesthetics. This permits the use of lower concentrations of both agents, minimizing local anesthetic-induced complications of motor blockade and the opioid-induced complications listed above. Epidural administration of opioids has become popular in the management of postoperative pain and for providing analgesia during labor and delivery. Lower systemic opioid levels are achieved with epidural opioids, leading to less placental transfer and less potential for respiratory depression of the newborn (Shnider and Levinson, 1987). Intrathecal ('spinal' anesthesia) administration of opioids as a single bolus also is popular for acute pain management. Chronic intrathecal infusions generally are reserved for use in chronic pain patients. Peripheral Analgesia As previously mentioned, opioid receptors on peripheral nerves have been shown to respond to locally applied opioids during inflammation (Stein, 1995). Peripheral analgesia permits the use of lower doses, applied locally, than those necessary to achieve a systemic effect. The effectiveness of this technique has been demonstrated in studies of postoperative pain (Stein et al., 1991). These studies also suggest that peripherally acting opioid compounds would be effective in other selected circumstances without entering the CNS to cause many undesirable side effects. Development of such compounds and expansion of clinical applications of this technique currently are active areas of research. Rectal Administration This route is an alternative for patients with difficulty swallowing
or other oral pathology and who prefer a less-invasive route than parenteral
(De Conno et al., 1995). This route is not well tolerated in most
children. Onset of action is seen within 10 minutes. In the Administration by Inhalation Preliminary studies have shown that opioids delivered by nebulizer can be an effective means of analgesic drug delivery (Worsley et al., 1990; Higgins et al., 1991). However, constant supervision is required when administering the drug, and variable delivery to the lungs can cause differences in therapeutic effect. In addition, possible environmental contamination is a concern. However, development of the inhaled route could provide a more convenient and cost-effective, adjunctive method of analgesic delivery for patients experiencing chronic pain. Oral Transmucosal Administration Opioids can be absorbed through the oral mucosa more rapidly than through the stomach. Bioavailability is greater due to avoidance of first pass metabolism, and lipophilic opioids are better absorbed by this route than are hydrophilic compounds such as morphine (Weinberg et al., 1988). A transmucosal delivery system that suspends fentanyl in a dissolvable matrix has been approved for clinical use (ACTIQ). Its primary indication is for treatment of breakthrough cancer pain (Ashburn et al., 1989). In this setting, transmucosal fentanyl relieves pain within 15 minutes, and patients easily can titrate the appropriate dose. Transmucosal fentanyl also has been studied as a premedicant for children. However, this technique has been largely abandoned due to a substantial incidence of undesirable side effects such as respiratory depression, sedation, nausea, vomiting, and pruritus. Transdermal or Iontophoretic Administration Transdermal fentanyl patches are approved for use with sustained pain. The opioid permeates the skin, and a 'depot' is established in the stratum corneum layer. Unlike other transdermal systems (i.e., transdermal scopolamine), anatomic position of the patch does not affect absorption. However, fever and external heat sources of heat (heating pads, hot baths) can increase absorption of fentanyl and potentially lead to an overdose (Rose et al., 1993). This modality is well suited for cancer pain treatment because of its ease of use, prolonged duration of action, and stable blood levels (Portenoy et al., 1993). It may take up to 12 hours to develop analgesia and up to 16 hours to observe full clinical effect. Plasma levels stabilize after two sequential patch applications, and these kinetics do not appear to change with repeated applications (Portenoy, 1993). However, there may be a great deal of variability in plasma levels after a given dose. The plasma half-life after patch removal is about 17 hours. Thus, if excessive sedation or respiratory depression is experienced, antagonist infusions may need to be maintained for an extended period (Payne, 1992). Dermatological side effects from the patches, such as rash and itching, usually are mild. Iontophoresis is the transport of soluble ions through the skin by using a mild electric current. This technique has been employed with morphine (Ashburn et al., 1992). Fentanyl and sufentanil have been chemically modified and applied by iontophoresis in rats (Thysman and Preat, 1993). Effective analgesia was achieved in less than 1 hour, suggesting that iontophoresis could be a promising modality for postoperative pain. It should be noted that increasing the applied current will increase drug delivery and could lead to overdose. However, unlike transdermal opioids, a drug reservoir does not build up in the skin, thus limiting the duration of both main and side effects. General Principles of Opioid Use Opioid analgesics provide symptomatic relief of pain, but the underlying disease remains. The physician must weigh the benefits of this relief against any potential risk to the patient, which may be quite different in an acute compared with a chronic disease. In acute problems, opioids will reduce the intensity of pain. However, physical signs (such as abdominal rigidity) will generally remain. Relief of pain also can facilitate history taking, examination, and the patient's ability to tolerate diagnostic procedures. Patients should not be evaluated inadequately because of the physician's unwillingness to prescribe analgesics, nor in most cases should analgesics be withheld for fear of obscuring the progression of underlying disease. The problems that arise in the relief of pain associated with chronic conditions are more complex. Repeated daily administration eventually will produce tolerance and some degree of physical dependence. The degree will depend on the particular drug, the frequency of administration, and the quantity administered. The decision to control any chronic symptom, especially pain, by the repeated administration of an opioid must be made carefully. When pain is due to chronic, nonmalignant disease, measures other than opioid drugs should be employed to relieve chronic pain if they are effective and available. Such measures include the use of nonsteroidal antiinflammatory agents, local nerve block, antidepressant drugs, electrical stimulation, acupuncture, hypnosis, or behavioral modification (see Foley, 1985). However, highly selected subpopulations of chronic nonmalignant pain patients can be adequately maintained on opioids for extended periods of time (Portenoy, 1990). In the usual doses, morphine-like drugs relieve suffering by altering the emotional component of the painful experience as well as by producing analgesia. Control of pain, especially chronic pain, must include attention to both psychological factors and the social impact of the illness that sometimes play dominant roles in determining the suffering experienced by the patient. In addition to emotional support, the physician also must consider the substantial variability in both the patient's capacity to tolerate pain and the response to opioids. As a result, some patients may require considerably more than the average dose of a drug to experience any relief from pain; others may require dosing at shorter intervals. Some clinicians, out of an exaggerated concern for the possibility of inducing addiction, tend to prescribe initial doses of opioids that are too small or given too infrequently to alleviate pain and then respond to the patient's continued complaints with an even more exaggerated concern about drug dependence, despite the high probability that the request for more drug is only the expected consequence of the inadequate dosage initially prescribed (see Sriwatanakul et al., 1983). It also is important to note that infants and children are probably more apt to receive inadequate treatment for pain than are adults due to communication difficulties, lack of familiarity with appropriate pain assessment methodologies, and inexperience with the use of strong opioids in children. If an illness or procedure causes pain for an adult, there is no reason to assume that it will produce less pain for a child (see Yaster and Deshpande, 1988). Pain of Terminal Illness and Cancer Pain Opioids are not indicated in all cases of terminal illness, but the analgesia, tranquility, and even the euphoria afforded by the use of opioids can make the final days far less distressing for the patient and family. Although physical dependence and tolerance may develop, this possibility should not in any way prevent physicians from fulfilling their primary obligation to ease the patient's discomfort. The physician should not wait until the pain becomes agonizing; no patient should ever wish for death because of a physician's reluctance to use adequate amounts of effective opioids. This may sometimes entail the regular use of opioid analgesics in substantial doses. Such patients, while they may be physically dependent, are not 'addicts' even though they may need large doses on a regular basis. Physical dependence is not equivalent to addiction (see Chapter 24: Drug Addiction and Drug Abuse). Most clinicians who are experienced in the management of chronic pain associated with malignant disease or terminal illness recommend that opioids be administered at sufficiently short, fixed intervals so that pain is continually under control and patients do not dread its return (Foley, 1993). Less drug is needed to prevent the recurrence of pain than to relieve it. Morphine remains the opioid of choice in most of these situations, and the route and dose should be adjusted to the needs of the individual patient. Many clinicians find that oral morphine is adequate in most situations. Sustained-release preparations of oral morphine are now available that can be administered at 8- to 12-hour intervals. Superior control of pain often can be achieved with fewer side effects using the same daily dose; a decrease in the fluctuation of plasma concentrations of morphine may be partially responsible. Constipation is an exceedingly common problem when opioids are used,
and the use of stool softeners and laxatives should be initiated early.
Amphetamines have demonstrable mood-elevating and analgesic effects and
enhance opioid-induced analgesia. However, not all terminal patients require
the euphoriant effects of amphetamine, and some experience side effects, such
as anorexia. Controlled studies demonstrate no superiority of oral heroin
over oral morphine. Similarly, after adjustment is made for potency,
parenteral heroin is not superior to morphine in terms of analgesia, effects
on mood, or side effects (see Sawynok, 1986). Although tolerance does
develop to oral opioids, many patients obtain relief from the same dosage for
weeks or months. In cases where one opioid loses effectiveness, switching to
another may provide better pain relief. 'Cross-tolerance' among
opioids exists, but, both clinically and experimentally, cross-tolerance
among related When opioids and other analgesics are no longer satisfactory, nerve block, chordotomy, or other types of neurosurgical intervention such as neurostimulation may be required if the nature of the disease permits. Epidural or intrathecal administration of opioids may be useful when administration of opioids by usual routes no longer yields adequate relief of pain (see above). This technique has been used with ambulatory patients over periods of weeks or months (see Gustafsson and Wiesenfeld-Hallin, 1988). Moreover, portable devices have been developed that permit the patient to control the parenteral administration of an opioid while remaining ambulatory (Kerr et al., 1988). These devices use a pump that infuses the drug from a reservoir at a rate that can be tailored to the needs of the patient, and they include mechanisms to limit dosage and/or allow the patient to self-administer an additional 'rescue' dose if there is a transient change in the intensity of pain. Nonanalgesic Therapeutic Uses of Opioids Dyspnea Morphine is used to alleviate the dyspnea of acute left ventricular failure and pulmonary edema, and the response to intravenous morphine may be dramatic. The mechanism underlying this relief still is not clear. It may involve an alteration of the patient's reaction to impaired respiratory function and an indirect reduction of the work of the heart due to reduced fear and apprehension. However, it is more probable that the major benefit is due to cardiovascular effects, such as decreased peripheral resistance and an increased capacity of the peripheral and splanchnic vascular compartments (see Vismara et al., 1976). Nitroglycerin, which also causes vasodilation, may be superior to morphine in this condition (see Hoffman and Reynolds, 1987). In patients with normal blood gases but severe breathlessness due to chronic obstruction of airflow ('pink puffers'), drocode (dihydrocodeine), 15 mg orally before exercise, reduces the feeling of breathlessness and increases exercise tolerance (Johnson et al., 1983). Opioids are relatively contraindicated in pulmonary edema due to respiratory irritants unless severe pain also is present; relative contraindications to the use of histamine-releasing opioids in asthma already have been discussed. Special Anesthesia High doses of morphine or other opioids have been used as the primary anesthetic agents in certain surgical procedures. Although respiration is so depressed that physical assistance is required, patients can retain consciousness (see Chapter 14: General Anesthetics). |
Prospectus
|
Great strides are being made in understanding
structure-function relationships between opioids and endogenous opioid
peptides and their receptors. The complex signaling mechanisms and neural
circuitry mediating both the salutary and undesirable effects of opioids also
are beginning to be understood. The recent discovery of new Dedication The authors would like to dedicate this chapter to the memory of Dr. Thomas F. Burks, colleague and friend, who had a major impact on the field of opioid pharmacology. Acknowledgment The authors wish to acknowledge Drs. Terry Reisine and Gavril Pasternak, authors of this chapter in the ninth edition of Goodman and Gilman's The Pharmacological Basis of Therapeutics, some of whose text has been retained in this edition. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1859
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2024 . All rights reserved