| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Pharmacological Treatment of Heart Failure
Overview
|
Heart failure is one of the most common
causes of death and disability in industrialized nations and is among the
syndromes most commonly encountered in clinical practice. Over 4.6 million
patients in the This chapter deals with drug therapy of heart failure due to systolic and/or diastolic ventricular dysfunction. Systolic dysfunction due to idiopathic dilated or ischemic cardiomyopathies usually is characterized by large, dilated ventricular chambers. Diastolic dysfunction due to long-standing hypertension, stenotic valvular disease, or a primary hypertrophic cardiomyopathy generally leads to thickened, poorly compliant ventricular walls with small ventricular volumes. In reality, many patients exhibit abnormal hemodynamics comprising significant degrees of both systolic and diastolic dysfunction. Treatment must therefore be tailored to the underlying pathophysiological process in the individual patient. The basic pharmacology of many of the classes of drugs described in
this chapter is discussed in detail in other chapters and cross-referenced in
the text. Therefore, the pharmacology of these agents is discussed here only
in the context of the treatment of heart failure. The main elements in the
pharmacotherapy of heart failure are angiotensin converting enzyme inhibitors
(seeChapter 31: Renin and Angiotensin) and |
Goals of Therapy
|
Relief of Symptoms A primary goal in the treatment of heart failure is the alleviation of symptoms, which, in turn, are a direct result of the underlying hemodynamic disorder. Intravascular volume expansion and elevated ventricular filling pressures result in systemic and pulmonary venous hypertension, which causes dyspnea on exertion and orthopnea. Reduced cardiac output results in fatigue and decreased exercise capacity. In the short term, symptomatic treatment is directed at improving
hemodynamic function through the use of drugs that increase cardiac output
and reduce ventricular filling pressures. In the patient hospitalized because
of severe symptoms of heart failure, rapid treatment may include the use of
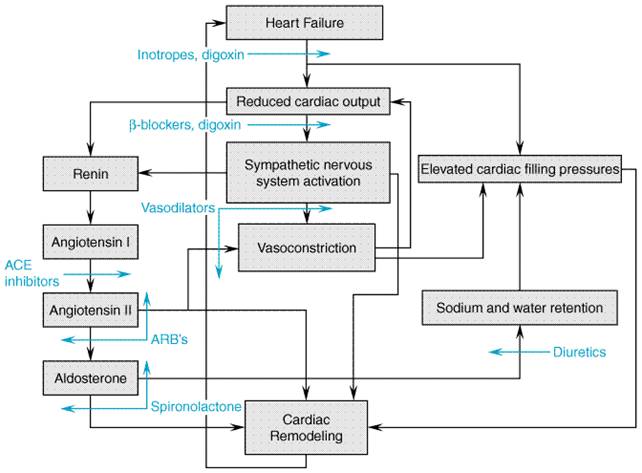
intravenous diuretics, positive inotropic agents (e.g., Myocardial Remodeling Even in the absence of recurrent damage to the heart, the severity of the underlying myocardial dysfunction often is progressive. This is due to ventricular 'remodeling,' a process that results in progressive maladaptive changes in the structure and function of the ventricle (Cohn, 1995). Therefore, a second major goal of therapy is to slow or prevent the progression of myocardial remodeling. Initially, myocardial dysfunction causes intravascular volume expansion and the activation of neurohormonal systems, particularly the sympathetic nervous and reninangiotensin systems. These primitive, compensatory responses defend the perfusion of vital organs by increasing left ventricular preload, stimulating myocardial contractility, and increasing arterial tone. However, with time each plays a role in the pathophysiology of the disease by promoting the progression of the underlying myocardial dysfunction. Expansion of the intravascular volume results in elevated ventricular filling pressures which increase ventricular wall stresses. Neurohormonal activation causes arterial and venous constriction which also leads to increased ventricular wall stresses. In addition, some neurohormones (e.g., norepinephrine and angiotensin) may act directly on the myocardium to promote remodeling by causing myocyte apoptosis, abnormal gene expression, and/or alterations in the extracellular matrix (Colucci and Braunwald, 2000). Drugs that reduce ventricular wall stresses (e.g.,
vasodilators) and/or inhibit the reninangiotensin system (e.g.,
angiotensin converting enzyme inhibitors) or the sympathetic nervous system (e.g.,
The pharmacotherapeutic management of patients with heart failure is described in two sections. The first describes the use of oral drugs in ambulatory patients. The second describes intravenously administered agents primarily used for the treatment of hospitalized patients |
Oral Drugs for the Management of Ambulatory Heart Failure
|
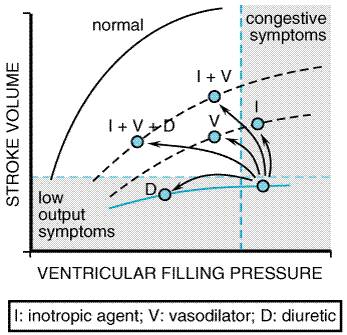
Diuretics For several decades, and especially following the introduction of 'loop' diuretics, this group of drugs has played a central role in the pharmacological management of the 'congestive' symptoms in heart failure. Diuretics are discussed in detail in Chapter 29: Diuretics. Therefore, only those aspects of their pharmacology that are relevant to the treatment of heart failure will be dealt with in this chapter. The importance of these drugs is due to the central role of the kidney as the target organ for many of the hemodynamic, hormonal, and autonomic nervous system changes that occur in response to a failing myocardium. The net effect of these changes is the retention of salt and water and expansion of the extracellular fluid volume, which serves in the short run to sustain cardiac output and tissue perfusion by allowing the heart to operate higher on its ventricular function (i.e., FrankStarling) curve (Figure 342). However, this response incurs the cost of higher end-diastolic filling pressures and increasing ventricular chamber dimensions and wall stress, which eventually limit any further increase in cardiac output and also result in pulmonary venous congestion and peripheral edema.
Diuretics act to reduce extracellular fluid volume and ventricular filling pressures (or 'preload') but usually do not cause a clinically important reduction in cardiac output, particularly in patients with advanced heart failure who have an increased left ventricular filling pressure, unless there has been a profound and sustained natriuresis that results in a rapid decline in intravascular volume. Despite the clear efficacy of diuretics in controlling congestive symptoms and improving exercise capacity, it is worth noting that, with the exception of low-dose spironolactone, the use of diuretics has not been demonstrated to improve survival in heart failure. Indeed, monotherapy with a diuretic may cause increased neurohormonal activation due to volume depletion, with potentially deleterious effects on the progression of heart failure. For this reason, it may be preferable to avoid the use of diuretics in the subset of patients who have mild heart failure without evidence of fluid retention. Dietary Sodium Restriction All patients with clinically significant ventricular dysfunction, regardless of whether or not they are symptomatic, should be advised to limit dietary intake of NaCl. Most patients will tolerate moderate reductions in salt intake (2 to 3 g/day total intake). More stringent salt restriction is seldom necessary and may be counterproductive in many heart-failure patients as it may lead to hyponatremia, hypokalemia, and a metabolic alkalosis due to chloride depletion when combined with loop diuretics, as well as loss of lean body mass due to reduced appetite. Of the loop diuretics currently available, only furosemide (LASIX), bumetanide (BUMEX), and torsemide (DEMADEX) are indicated in the treatment of most patients with heart failure. Due to the increased risk of ototoxicity, ethacrynic acid should be reserved for patients who are allergic to sulfonamides or who have developed interstitial nephritis on alternative drugs. The bioavailability of orally administered furosemide ranges from 40% to 70%, and adjustments in dosage may be required before it is deemed ineffective. In contrast, bumetanide and torsemide have oral bioavailabilities of more than 80% and provide more consistent absorption, albeit at a considerably greater cost. Furosemide and bumetanide are short-acting drugs. Avid renal Na+ retention by all nephron segments following a decline in renal tubular diuretic levels can limit or prevent a negative Na+ balance. In many patients with heart failure, this necessitates the use of two or more daily doses of these diuretics to induce and sustain a negative salt balance. This is an acceptable strategy for outpatient management of heart failure, provided that there is adequate monitoring of daily weights and blood electrolyte levels. Thiazide Diuretics The principal site of action of the thiazide diuretics is now known to be the Na+Cl cotransporter (seeChapter 29: Diuretics) present in renal tubular epithelial cells in the distal convoluted tubule. The thiazide diuretics generally are useful as single drugs for the therapy of volume retention only in patients with relatively mild heart failure, largely because their site of action in the distal nephron permits rapid adjustment of water and solute absorption by other, more proximal nephron segments. Thiazide diuretics also are ineffective at glomerular filtration rates below 30 ml/minute. However, these drugs exhibit true synergism with loop diuretics (i.e., a natriuresis that is greater than the sum of either class of drugs given individually). This is useful when patients become refractory to loop diuretics (see'Diuretic Resistance in Heart Failure,' below). K+-Sparing Diuretics K+-sparing diuretics (seeChapter 29: Diuretics) are divided into those agents that inhibit apical membrane Na+ conductance channels in epithelial cells of the collecting duct (e.g., amiloride, triamterene) and aldosterone antagonists that also have their principal pharmacological effect in the collecting duct (e.g., spironolactone, canrenone). Although these agents generally are not effective as diuretic agents when used alone, they may be useful in limiting renal K+ and Mg2+ wasting and/or in augmenting the response to other classes of diuretics. As discussed below, there is evidence that a low dose of spironolactone may improve survival in patients with advanced symptoms of heart failure, apparently via a mechanism independent of diuresis (Pitt et al., 1999). Use of Diuretics in Clinical Practice The majority of patients with heart failure will require the chronic administration of a loop diuretic to maintain euvolemia. In patients with fluid retention, furosemide typically is started at a dose of 40 mg once or twice per day, and the dosage is increased until an adequate diuresis (increased urine output and weight loss of 0.5 to 1.5 kg daily) is achieved. A larger initial dose may be required if there is significant renal impairment, or in severe heart failure. Serum electrolytes and renal function should be monitored, especially if there is preexisting renal insufficiency or a brisk diuresis is desirable in a severely symptomatic patient. Once fluid retention has resolved, the dose of diuretic should be reduced to the minimum necessary to maintain euvolemia. Electrolyte abnormalities or worsening azotemia may supervene before euvolemia is achieved. Hypokalemia may occur and may be corrected by potassium supplementation or addition of a potassium-sparing diuretic. In general, diuresis should be slowed only if azotemia or renal impairment become progressive or the patient is symptomatic. Diuretic Resistance in Heart Failure The response to diuretics frequently is impaired in heart failure. Following prolonged administration of a loop diuretic, a process of adaptation occurs in which there is a compensatory increase in sodium reabsorption in the distal nephron and blunting of net sodium and water loss. Furthermore, while there may be a brisk response following a single dose of diuretic, a compensatory increase in sodium reabsorption for the remaining part of the 24-hour period may prevent effective diuresis. Patients who have impaired renal function typically require higher doses of diuretic to ensure adequate delivery of the diuretic to its site of action. A poor response to diuretics also may be due to edema and decreased motility of the bowel wall as well as to reduced splanchnic blood flow. This may cause slowed absorption and a delay in the peak effect of orally administered diuretics, although the total amount absorbed usually is unchanged. The more common causes of diuretic resistance are listed in Table 341. It often is difficult to determine clinically whether an increasing diuretic requirement is due to intravascular volume depletion following aggressive diuretic and vasodilator therapy or to a decrease in cardiac output and blood pressure due to the underlying cardiac failure. Invasive monitoring to determine the left ventricular filling pressure may be required to make this distinction. However, a more marked decline in urea clearance than in creatinine clearance (resulting in an increase in the BUN to creatinine ratio) suggests intravascular volume depletion. Vasodilators commonly employed as 'unloading' agents in heart failure may reduce renal blood flow despite an increase in cardiac output, thereby reducing diuretic effectiveness. Also, since some patients with heart failure also may have renal arterial atherosclerotic disease, vasodilator therapy may lower renal perfusion pressure below that necessary to maintain normal autoregulation and glomerular filtration. The caveats about vasodilators also apply to ACE inhibitors and to angiotensin II type 1 receptor (i.e., AT1 receptor) antagonists (seeChapter 31: Renin and Angiotensin). However, because of the unique role of angiotensin II as an intrarenal signaling autocoid, these drugs can either augment the effectiveness of diuretics by mechanisms independent of their ability to reduce systemic vascular resistance or diminish their effectiveness by reducing the transglomerular perfusion pressure to the point that the glomerular filtration rate declines abruptly. The latter response is observed most commonly in patients with decreased renal arterial perfusion pressure, due either to renal artery stenosis and/or a limited cardiac output, for whom a high angiotensin IImediated glomerular efferent arteriolar tone is necessary to maintain glomerular filtration. This cause of diuretic resistance generally is accompanied by a decline in creatinine clearance and should be distinguished from the more modest, limited rise in serum creatinine levels and improved responsiveness to diuretics that commonly accompany ACE-inhibitor administration. Diuretic resistance due primarily to poor cardiac function generally improves when cardiac output is increased by the use of positive inotropic agents (e.g., dobutamine) or vasodilators. Decreased responsiveness to loop diuretics in patients otherwise receiving optimal medical management should be managed initially by increasing the frequency of doses and by more stringent dietary salt restriction. If this is ineffective, a thiazide diuretic (e.g., hydrochlorothiazide or metolazone) administered with the loop diuretic often is effective (Ellison, 1991). However, this diuretic combination can result in an unpredictable and at times excessive diuresis, which can cause intravascular volume depletion and renal K+ wasting; the combination therefore should be used cautiously. Spironolactone also may be effective in these patients when combined with a loop diuretic. For a detailed discussion on the subject of diuretic resistance, the reader is referred to a recent comprehensive review (Ellison, 1999). Metabolic Consequences of Diuretic Therapy The side effects of diuretic agents are discussed in Chapter 29: Diuretics and in recent overviews of diuretic use (e.g., Brater, 1998). With regard to diuretic use in heart failure, the most important adverse sequelae of diuretics are electrolyte abnormalities, including hyponatremia, hypokalemia, and hypochloremic metabolic alkalosis. The clinical importance, or even the existence, of significant Mg2+ deficiency with chronic diuretic use remains controversial (Bigger, 1994; Davies and Fraser, 1993). Both hypokalemia and renal Mg2+ wasting can be limited by administration of oral KCl supplements or a K+-sparing diuretic. Aldosterone Antagonists One of the principal features of heart failure is marked activation of the renin-angiotensin-aldosterone system with elevation of the plasma aldosterone concentration to as much as 20 times the normal level. As mentioned above, when used alone spironolactone exerts only a very weak diuretic effect in patients with heart failure. However, aldosterone has a range of biological effects in addition to salt retention (seeTable 342), and it has been suggested that antagonism of aldosterone, per se, may be beneficial in patients with heart failure. For a review of the subject of aldosterone and spironolactone in heart failure, see the recent review by Struthers (1999). Clinical Use of Spironolactone in Heart Failure The Randomized Aldactone Evaluation Study (RALES) randomized patients
with moderate to severe heart failure [New York Heart Association (NYHA)
class III to IV] to receive 25 mg daily of spironolactone or placebo in
addition to conventional therapy, which included, in the large majority of
patients, an ACE inhibitor (Pitt, 1999). Patients with serum creatinine
concentrations greater than 2.5 mg/dl (221 The RALES trial suggests that the beneficial effects of spironolactone are additive to those of ACE inhibitors, and its use should be considered in patients with NYHA class III to IV heart failure. However, caution should be exercised in its use when significant renal impairment is present. Treatment should be initiated at a dose of 12.5 or 25 mg daily. Higher doses should be avoided, as they may lead to hyperkalemia, particularly in patients receiving an ACE inhibitor. Serum potassium levels and electrolytes should be checked after initiation of treatment, and vigilance should be exercised for potential drug interactions and medical disorders that may cause elevations in serum potassium concentration (e.g., potassium supplements, ACE inhibitors, and worsening renal function). Vasodilators Although several classes of drugs exhibit vasodilator activity and may
improve symptoms in heart failure (Table 343), only ACE inhibitors
and the hydralazineisosorbide dinitrate combination have been shown
to improve survival in prospective randomized trials. Some classes of drugs,
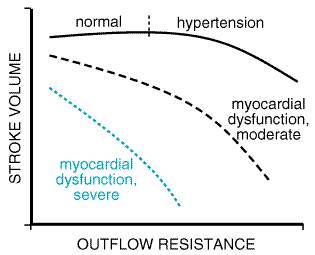
such as The rationale for the use of drugs with vasodilatory activity in heart failure grew out of experience with parenteral phentolamine and nitroprusside in patients with severe heart failure. Cohn and Franciosa, in an influential article in 1977, reviewed the evidence supporting this approach. Studies of ACE inhibitors in the following decade showed that these drugs generally were well tolerated and effective in improving symptoms in heart failure, while two randomized, prospective trials verified the effectiveness of an isosorbide dinitratehydralazine combination (Cohn et al., 1986) and enalapril (CONSENSUS, 1987) in reducing mortality in patients with heart failure. Subsequent clinical trials have reinforced the results of these two trials and have provided evidence supporting expanded indications for the use of ACE inhibitors to patients with ventricular dysfunction but without overt symptoms of heart failure (see below). Principles of Vasodilator Therapy The principles of vasodilator therapy in heart failure are reviewed in detail in texts of cardiovascular medicine and physiology (e.g., Smith et al., 1997). Briefly, the hemodynamic responses to heart failure are similar in some respects to those that accompany a fall in blood pressure due to hypovolemia; they include tachycardia and an increase in venous and arterial vasoconstriction, with shunting of blood toward the thorax and brain and away from the periphery and splanchnic and renal vascular beds. While this provides a clear evolutionary advantage to the organism to survive dehydration or hemorrhage, it is maladaptive and deleterious in chronic heart failure. The concepts of preload and afterload reduction provide a convenient framework in which to address treatment options in heart failure, which, in many respects, attempt to overcome these inappropriate compensatory hemodynamic responses. While this discussion focuses on heart failure due to left ventricular dysfunction, the general principles of preload and afterload reduction are applicable to failure of either ventricle despite differences in the specific drugs or other forms of therapy that can be employed. Although drugs may be classified as either 'arterial' or 'venous' vasodilators, most vasodilators exhibit activity on both vascular beds. Also, classes of vasodilators differ in the specific arterial beds that are affected. This has important implications in preserving, for example, renal blood flow and diuretic effectiveness and may explain, in part, the superior efficacy of certain classes of vasodilator agents in heart failure. Preload Reduction The principle of preload reduction can be expressed in the form of the FrankStarling relationship illustrated in Figure 342. In early heart failure, increases in intraventricular volume and pressure, as well as heart rate, compensate for a decline in ventricular systolic performance due to underlying cardiac disease. In more advanced heart failure, there may be little or no further augmentation of stroke volume with increasing filling pressures (i.e., a 'flat FrankStarling curve'), while the transmission of increased pressure into the pulmonary and systemic venous beds produces congestive symptoms. This usually is accompanied by worsening myocardial energetics due to an increase in ventricular wall stress and a decrease in diastolic coronary blood flow. Agents that reduce ventricular filling pressures by decreasing intravascular volume (e.g., diuretics) or by increasing venous capacitance (e.g., venodilators) decrease pulmonary venous congestion and may improve myocardial metabolism with minimal effects on stroke volume and cardiac output. While these measures clearly improve symptoms due to systolic ventricular dysfunction, they also benefit patients with congestive symptoms due to impaired diastolic compliance (i.e., 'diastolic dysfunction'), whether due to ischemia or structural changes in the myocardium. However, patients with poorly compliant hypertrophied ventricles due, for example, to aortic stenosis, often require elevated end-diastolic filling pressures to support an adequate forward stroke volume. An excessive decrease in preload may markedly reduce cardiac output in these patients. Afterload Reduction The importance of systemic arterial vasodilation in improving cardiac hemodynamics in heart failure was succinctly described by Cohn and Franciosa (1977). Afterload, the sum of forces opposing ventricular emptying during systole, is dependent on aortic and aortic outflow tract (including valvular) impedance, systemic vascular resistance, ventricularvascular coupling (i.e., the harmonics of reflected arterial pressure waves during systole), and the volume of blood in the ventricle at the initiation of systole. Hypertrophy of ventricular muscle is a compensatory mechanism that reduces wall stress and preserves ventricular systolic function despite a primary abnormality in one or more of the determinants of afterload (e.g., a stenotic aortic valve). In the failing heart, as illustrated in Figure 343, any reduction in ventricular wall stress during systole, whether achieved by corrective surgery, intraaortic balloon counterpulsation, or vasodilator drugs, results in improved systolic contractile function. In addition, any reduction in aortic or arterial determinants of afterload will improve forward stroke volume and improve signs and symptoms of mitral regurgitation, which is often present in patients with severe heart failure due to systolic dysfunction, even in the absence of primary disease of the mitral valve.
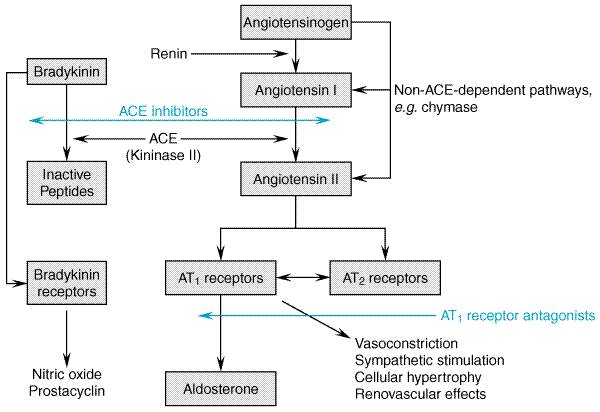
Inhibition of the ReninAngiotensin System The reninangiotensin system has a central role in the pathophysiology of heart failure (for detailed description of the reninangiotensin system, seeChapter 31: Renin and Angiotensin). Angiotensinogen is cleaved by kidney-derived renin to form the decapeptide angiotensin I; ACE converts angiotensin I to the octapeptide angiotensin II (seeFigure 344). Angiotensin II is a potent arterial vasoconstrictor which promotes sodium and water retention through its role in the regulation of renal hemodynamics and release from the adrenal cortex of aldosterone. Additionally, angiotensin II potentiates catecholamine release, is arrhythmogenic, promotes vascular hyperplasia and pathologic myocardial hypertrophy, and stimulates myocyte death. These actions of angiotensin II contribute to the pathophysiology of heart failure and, in many cases, to pathological remodeling of the myocardium leading to disease progression.
ACE inhibitors have been shown to suppress angiotensin II and aldosterone production, decrease sympathetic nervous system activity, and potentiate the effects of diuretics in heart failure. However, angiotensin II levels frequently return to baseline values following chronic treatment with ACE inhibitors (Juillerat et al., 1990). This is believed to be due to production of angiotensin II through non-ACE-dependent pathwaysfor example, through the action of chymase, a tissue protease. The sustained clinical effectiveness of ACE inhibitors despite failure to maintain angiotensin II suppression has raised the possibility that there may be additional or alternate mechanisms by which ACE inhibitors work in heart failure. ACE is identical to kininase II, which degrades bradykinin and other kinins; bradykinin stimulates production of nitric oxide, cyclic GMP and vasoactive prostaglandins which result in vasodilation and oppose the effects of angiotensin II on vascular- and myocardial-cell growth. Thus, it has been suggested that increased levels of bradykinin as a result of ACE inhibition may play an important role in the hemodynamic and antiremodeling effects of ACE inhibitors. ACE inhibitors cause both arterial and venous dilation. There are reductions in systemic and pulmonary arterial resistances. Mean arterial pressure may be unchanged or decrease, but heart rate usually is unchanged, even when there is a decrease in systemic pressure, perhaps due to a decrease in sympathetic nervous system activity. The decrease in left ventricular afterload results in an increase in cardiac output due to increases in stroke volume and ejection fraction. Venodilation results in decreases in right and left heart filling pressures and end-diastolic volumes. An alternative means of inhibiting the reninangiotensin system is through inhibition of angiotensin receptors. Most of the known clinical actions of angiotensin II, including its deleterious effects in heart failure, are mediated through the type 1 angiotensin receptor (AT1). Type 2 angiotensin receptors (AT2) also are present throughout the cardiovascular system, where they are believed to counterbalance the biological effects of AT1-receptor stimulation. Due to their more distal site of action, AT1 angiotensin-receptor blockers (ARBs) theoretically may allow more complete interruption of angiotensin's actions than can be obtained with ACE inhibitors. Furthermore, since AT1-receptor blockade has been associated with a compensatory increase in angiotensin II levels, AT2-receptor stimulation is increased, potentially enhancing the beneficial effects of ARBs. On the other hand, a theoretical drawback of the ARBs is their failure to increase bradykinin production. The relative merits of these possible mechanisms of action remain to be seen, as does the value of combined therapy with an ACE inhibitor and an ARB, which has the potential to provide more complete reninangiotensin system blockade while increasing bradykinin production. Angiotensin Converting Enzyme Inhibitors The first orally active ACE inhibitor, captopril (CAPOTEN), was introduced in 1977, and currently five other ACE inhibitorsenalapril (VASOTEC), ramipril (ALTACE), lisinopril (PRINIVIL ZESTRIL), quinapril (ACCUPRIL), and fosinopril (MONOPRIL), are approved by the U.S. Food and Drug Administration (FDA) for the treatment of heart failure. ACE inhibitors are now indicated for the treatment of heart failure of any severity, including asymptomatic left ventricular dysfunction. ACE-inhibition therapy should be initiated at a low dose (e.g., 6.25 mg of captopril or 5 mg of lisinopril), as some patients may experience an abrupt drop in blood pressure, particularly if they are volume depleted. Unacceptable hypotension usually can be reversed by intravascular volume expansion, although this may be counterproductive in patients with symptomatic heart failure. ACE inhibitor doses are customarily titrated upwards over several days in hospitalized patients or a few weeks in ambulatory patients, with careful observation of blood pressure, serum electrolytes, and serum creatinine levels. There is no precisely defined relationship between dose and long-term clinical effectiveness of these drugs. The target doses of these drugs in several large prospective trials in which a positive effect was demonstrated on mortality and other endpoints were 50 mg of captopril three times a day (Pfeffer et al., 1992); 10 mg of enalapril twice daily (SOLVD Investigators, 1991; Cohn et al., 1991); 10 mg of lisinopril once daily (GISSI-3 Investigators, 1994); or 5 mg twice daily of ramipril (AIRE Study Investigators, 1993). The question of optimal dosage of ACE inhibitor was addressed in the Assessment of Treatment with Lisinopril and Survival (ATLAS) study (Packer, 1999). High-dose lisinopril (32.5 or 35 mg) reduced the combined endpoint of mortality and hospitalization when compared to a low dose (2.5 or 5 mg). Based on the available evidence, the initial dosage of an ACE inhibitor should be titrated to the dosage that was shown to be of benefit in the major heart-failure trials. In patients who have not achieved an adequate clinical response, further uptitration to higher doses, as tolerated, may be of value. In patients with heart failure and reduced renal blood flow, ACE inhibitors, unlike other vasodilators, limit the kidney's ability to autoregulate glomerular perfusion pressure due to their selective effects on efferent arteriolar tone. If this occurs, the dose of ACE inhibitor should be reduced or another class of vasodilator added or substituted. Rarely, worsening of renal function following initiation of therapy with an ACE inhibitor will be due to the presence of bilateral renal artery stenosis. Another class of vasodilator should be substituted if this occurs. Likewise, angioneurotic edema secondary to ACE inhibitors should prompt immediate cessation of therapy. A small rise in serum potassium levels occurs frequently with ACE inhibitors; this may infrequently be substantial, especially in patients with renal impairment. Mild hyperkalemia is best managed by institution of a low potassium diet, but may require adjustment of dosage. A troublesome cough may occur, believed related to the effects of bradykinin. Substitution of an ARB often alleviates this problem. Effect of ACE Inhibitors on Survival in Heart Failure A number of placebo-controlled trials have demonstrated that ACE inhibitors improve survival in patients with overt heart failure due to systolic ventricular dysfunction regardless of the etiology or severity of symptoms. The Cooperative Northern Scandinavian Enalapril Survival Study (CONSENSUS, 1987) demonstrated a 40% reduction in mortality after 6 months in patients with severe heart failure randomized to enalapril rather than placebo. These results were confirmed by a 16% reduction in mortality in the treatment arm of the Studies On Left Ventricular Dysfunction (SOLVD Investigators, 1991) trial, in which patients with symptomatic mild to moderate heart failure and left ventricular ejection fractions less than 35% were randomized to receive either enalapril or placebo. The second Veterans Administration Cooperative VasodilatorHeart Failure Trial (V-HeFT II; Cohn et al., 1991) showed a small but clear survival benefit in patients with mild to moderate heart failure who were randomized to enalapril rather than the combination of hydralazine and isosorbide dinitrate. A smaller randomized trial comparing captopril to hydralazine and isosorbide dinitrate in patients with moderate to severe heart failure also demonstrated a significant survival advantage to patients receiving the ACE inhibitor (Fonarow et al., 1992). These data convinced many clinicians that ACE inhibitors clearly improved survival of patients with symptomatic heart failure. The prevention arm of the SOLVD trial subsequently examined whether or not asymptomatic patients with left ventricular systolic dysfunction also derive a survival benefit (SOLVD Investigators, 1992). Although this study failed to demonstrate a statistically significant reduction in mortality among enalapril-treated patients, there was a significant (29%) reduction in the combined end point of the development of symptomatic heart failure and death due to any cause. ACE inhibitors also may prevent the development of clinically significant ventricular dysfunction and mortality following myocardial infarction. The Survival And Ventricular Enlargement trial (SAVE; Pfeffer et al., 1992), which examined patients with recent, acute anterior myocardial infarction and ejection fractions of 40% or less, showed a 20% reduction in mortality and a 36% reduction in the rate of progression to severe heart failure in the captopril-treated group after 12 months of follow-up. Both the SOLVD trials (Konstam et al., 1992) and the SAVE trial (St. John Sutton et al., 1994) also demonstrated that enalapril and captopril, respectively, markedly reduced or prevented the increases in left ventricular end-diastolic and end-systolic volumes and decline in ejection fraction observed in patients randomized to receive placebo. The Acute Infarction Ramipril Efficacy trial (AIRE Investigators, 1993), which had a study design similar to that of the SAVE trial, also demonstrated a significant (27%) reduction in mortality in the ACE inhibitortreated group. These studies attest to the safety and efficacy of ACE-inhibitor therapy initiated early in the postinfarct period regardless of whether clinically significant left ventricular dysfunction is present at the time of randomization. Angiotensin II Receptor Antagonists Six orally active ARBs now have been approved for the treatment of hypertension, but none has been approved for use in heart failure. ARBs are discussed in detail in Chapter 31: Renin and Angiotensin and a recent review (Burnier and Brunner, 2000). In patients with heart failure, ARBs exert hemodynamic effects similar to those of the ACE inhibitors; however, there is much less information about their effects on long-term outcomes such as hospitalization and survival. The Evaluation of Losartan in the Elderly (ELITE) trial was primarily designed to examine the effects of the ARB losartan (COZAAR) on renal function in elderly patients with heart failure (Pitt et al., 1997). Patients were randomized to receive either the ACE inhibitor captopril, 50 mg three times daily, or losartan, 50 mg once daily; there were no significant differences in renal function between the two groups, but an unexpected reduction in mortality and hospitalizations from heart failure was noted in the losartan group. This was followed by the much larger ELITE II study which, like ELITE I, compared losartan and captopril in elderly patients, but had mortality as the primary end point (Pitt et al., 2000). In contrast to the findings in ELITE I, no significant difference in outcome was noted between the treatment groups. However, treatment with the ARB was better tolerated and associated with fewer adverse effects than treatment with the ACE inhibitor. Thus, ELITE II failed to confirm the superiority of an ARB over an ACE inhibitor. In addition, this study was not designed to test the equivalency of these two drug classes, and its interpretation has been confounded by concern that the dosage of losartan studied was not sufficient to achieve adequate blockade of AT1 receptors. Thus, until further data are available, the much larger body of data showing the benefits of ACE inhibitors in heart failure supports their routine use as first line agents. Conversely, although the present data do not allow the conclusion that ARBs are equivalent to ACE inhibitors, it appears reasonable to use ARBs as an alternative in patients intolerant to ACE inhibitors. Large trials are in progress that should provide more definitive data regarding the relative roles of ACE inhibitors and ARBs in the treatment of heart failure. The use of ACE inhibitors and ARBs in combination offers the interesting possibility of additive therapeutic effects due to their different modes of action on the reninangiotensin system. Preliminary results suggest that combined therapy with the ARB candesartan and the ACE inhibitor enalapril had favorable effects on hemodynamics, ventricular remodeling, and neurohormonal profile compared to therapy with either agent alone (McKelvie et al., 1999). Again, more definitive data are required before combination therapy is routinely applied in clinical practice. Nitrovasodilators Nitrovasodilators are among the oldest and most widely used vasodilators in clinical practice. The mechanism underlying the ability of these drugs to activate soluble guanylyl cyclase and relax vascular smooth muscle has become apparent only in the past 15 years. These drugs mimic the activity of nitric oxide (NO), an intracellular and paracrine signaling autocoid that is formed by the conversion of arginine to citrulline mediated by a family of enzymes termed nitric oxide synthases. This family of enzymes is found in endothelial and smooth muscle cells throughout the vasculature as well as in many other cell types. The basis for the differential sensitivity of selected regions of the vasculature to specific nitrovasodilators (e.g., the sensitivity of the epicardial coronary arteries to nitroglycerin, for example) remains controversial. Unlike nitroprusside, which is spontaneously converted to nitric oxide by reducing agents such as glutathione, nitroglycerin and other organic nitrates undergo a more complex enzymatic biotransformation to nitric oxide or bioactive S-nitrosothiols. The activities of specific enzyme(s) and cofactors required for this biotransformation, while not yet clearly identified, appear to differ within the vascular beds among organs and at different levels of the vasculature within an organ (Kelly and Smith, 1996). The basic pharmacology of the organic nitrates is discussed in Chapter 32: Drugs Used for the Treatment of Myocardial Ischemia. Organic Nitrates Organic nitrate preparations, most commonly isosorbide dinitrate (ISORDIL, DILATRATE, SORBITRATE), intravenous nitroglycerin, and nitroglycerin ointment, sublingual tablets, transdermal patch, and lingual spray, are relatively safe and effective agents in reducing ventricular filling pressures in acute as well as chronic congestive heart failure. The predominant effect at conventional doses is preload reduction due to an increase in peripheral venous capacitance. Nitrates also will cause a decline in pulmonary and systemic vascular resistance, particularly at higher doses, although this response is less marked and less predictable than with nitroprusside. Due to their relatively selective vasodilating effects on the epicardial coronary vasculature, these drugs may enhance systolic and diastolic ventricular function by increasing coronary flow in patients with underlying ischemia. Isosorbide dinitrate has been shown to be more effective than placebo
in improving exercise capacity and in reducing symptoms when administered
chronically to heart-failure patients. However, limited effects on systemic
vascular resistance and the problem of pharmacological tolerance have
restricted the use of organic nitrates as single agents in the
pharmacotherapy of heart failure. In a number of smaller trials, isosorbide
dinitrate has been shown to increase the clinical effectiveness of other
vasodilators such as hydralazine, resulting in a sustained improvement in
hemodynamics that exceeded those of either drug given alone. Importantly, the
combination of isosorbide dinitrate (20 mg four times daily) and hydralazine
in the V-HeFT I trial reduced overall mortality compared to either placebo or
the Nitrate tolerance limits the long-term effectiveness of these drugs when administered throughout the day for heart-failure symptoms. Blood levels of these drugs should be permitted to fall to negligible levels for at least 6 to 8 hours each day. The timing of nitrate withdrawal (e.g., removal of a transdermal nitroglycerin patch or skipping a dose of isosorbide dinitrate) can be adjusted to the patient's symptoms. Patients with recurrent orthopnea or paroxysmal nocturnal dyspnea, for example, would probably benefit most by using nitrates at night. Orally bioavailable compounds containing sulfhydryl groups, such as N-acetylcysteine, may diminish tolerance to the hemodynamic effects of nitrates in heart failure (Mehra et al., 1994). Likewise, hydralazine may decrease nitrate tolerance by a strong antioxidant effect reducing superoxide formation (which reacts rapidly with nitric oxide) and hence increasing bioavailability of nitric oxide (Gogia et al., 1995). Hydralazine The vasodilator activity of hydralazine (APRESOLINE) is not mediated by any known neural or hormonal agent, and its cellular mechanism of action in vascular smooth muscle remains poorly understood. Hydralazine is an effective antihypertensive drug (Chapter 33: Antihypertensive Agents and the Drug Therapy of Hypertension), particularly when combined with other agents that blunt compensatory increases in sympathetic tone and salt and water retention. In heart failure, hydralazine reduces right and left ventricular afterload by reducing systemic and pulmonary vascular resistance. This results in an augmentation of forward stroke volume and a reduction in ventricular systolic wall stress and regurgitant fraction in mitral insufficiency. Hydralazine also appears to have moderate 'direct' positive inotropic activity in cardiac muscle unrelated to afterload reduction. Hydralazine has minimal effects on venous capacitance and therefore is most effective when combined with agents with venodilating activity (e.g., organic nitrates). Importantly, hydralazine is effective in reducing renal vascular resistance and in increasing renal blood flow to a greater degree than are most other vasodilators, with the exception of ACE inhibitors. Therefore, hydralazine may be useful in heart-failure patients with renal dysfunction who cannot tolerate an ACE inhibitor. The combination of hydralazine (300 mg/day) and isosorbide dinitrate
was less effective than enalapril in reducing mortality in heart-failure
patients in the V-HeFT II trial (Cohn et al., 1991), although this
combination of agents did increase survival compared to placebo or the Side effects that may necessitate dose adjustment or withdrawal of hydralazine are common. Twenty percent of patients in the V-HeFT I trial (Cohn et al., 1986) complained of symptoms that could have been related to hydralazine, although the most common complaints, headache and dizziness, also could have been due to the concomitantly administered nitrates. Usually, the symptoms diminish with time or respond to a reduction in dose. The oral bioavailability and pharmacokinetics of elimination of hydralazine do not appear to be importantly affected by heart failure unless there is severe hepatic congestion or hypoperfusion. Intravenous hydralazine is available but provides little practical advantage over oral formulations except for urgent use during pregnancy, in which relative contraindications exist for most other vasodilators. Hydralazine is typically started at a dose of 10 to 25 mg three times a day and the dosage titrated to 100 mg three times a day over several days as clinical needs dictate and side effects allow. Ca2+ Channel Antagonists The Ca2+ channel antagonists are effective arterial vasodilators that have been widely used to treat hypertension (seeChapter 33: Antihypertensive Agents and the Drug Therapy of Hypertension). Although these agents offer theoretical advantages in the management of heart failure, the clinical experience has been disappointing. The use of the first-generation Ca2+ channel antagonists (verapamil, diltiazem, nifedipine) has not been shown to produce sustained improvement in symptoms in patients with predominant systolic ventricular dysfunction. Indeed, these drugs may worsen symptoms and increase mortality in patients with systolic dysfunction, including patients with heart failure due to ischemic disease (Elkayam et al., 1993). The reason for this adverse effect of Ca2+ channel blockers in heart failure is unclear, but may be related to their known negative inotropic effects or to reflex neurohumoral activation. Amlodipine (NORVASC) and felodipine (PLENDIL) are second-generation dihydropyridines with greater vascular selectivity, and hence, fewer negative inotropic effects than the first-generation agents. In the Prospective Randomized Amlodipine Survival Evaluation Study (PRAISE), 1153 patients with severe heart failure were randomized to receive amlodipine (up to 10 mg daily) or placebo (Packer et al., 1996a). A trend toward a decrease in mortality was noted in the amlodipine group, with a more pronounced survival benefit in the subgroup of patients with nonischemic cardiomyopathy. Therefore, PRAISE II further evaluated amlodipine in patients with nonischemic cardiomyopathy; preliminary results of this study, however, do not suggest a survival benefit with amlodipine, but confirmed its safety in this patient population. Similarly, felodipine was shown to have a neutral effect on survival and exercise tolerance in the third Vasodilator-Heart Failure Trial (V-HeFT III) (Cohn et al., 1997). The aforementioned Ca2+ channel antagonists act on the voltage-sensitive L-type channel. In contrast, mibefradil is a Ca2+ channel antagonist that is selective for the non-voltage-regulated, T-type channel. Although this agent does not appear to have clinically significant negative inotropic activity, its use was associated with an increased risk of death (believed secondary to major adverse drug interactions) that prompted its withdrawal from the market. The available clinical evidence does not support the routine use of Ca2+ channel antagonists as first-line therapy in patients with heart failure. However, the use of amlodipine or felodipine may be considered in certain circumstances in which additional control of blood pressure or afterload reduction is required, or if other vasodilators (e.g., ACE inhibitors, angiotensin receptor antagonists or hydralazine) are contraindicated or poorly tolerated. In contrast to results in patients with predominant systolic dysfunction, Ca2+ channel antagonists appear to be useful agents for the treatment of heart failure due predominantly to diastolic dysfunction, such as hypertensive or idiopathic hypertrophic cardiomyopathies. By slowing heart rate, which is an important determinant of diastolic filling time, verapamil and diltiazem may facilitate diastolic relaxation and lower diastolic filling pressures. These agents also can be useful in the acute management of patients in heart failure due to most supraventricular tachyarrhythmias in the absence of severe right or left ventricular systolic dysfunction, or a known or suspected extranodal atrioventricular accessory pathway (Wolff-Parkinson-White syndrome). Other Vasodilators As noted in Table 343, other vasodilator drugs are effective in reducing ventricular preload and afterload and improving symptoms in heart failure. None of these agents, however, has been shown to improve survival in heart-failure patients. Their use should be restricted to the treatment of patients who are intolerant to, or not adequately treated by, the agents discussed above. For the treatment of acute or chronic decompensated heart failure refractory to standard drug regimens and not complicated by significant aortic insufficiency, a mechanical aortic counterpulsation device (i.e., intraaortic balloon pump) often provides the most effective short-term means to reduce left ventricular afterload and directly increase cardiac output.
The pharmacology of
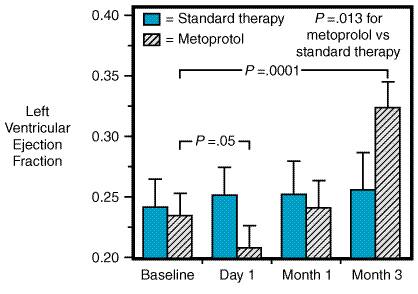
Early Outcome Trials The Metoprolol in Dilated Cardiomyopathy (MDC) trial (Waagstein et
al., 1993) was a multicenter, prospective, randomized trial that examined
metoprolol versus placebo in 383 patients with mild to moderate idiopathic
dilated cardiomyopathy (i.e., patients with clinically evident
coronary artery disease or active myocarditis were excluded) who already were
receiving optimal medical management including ACE inhibitors. Although there
was no difference in mortality at 12 months of follow-up between the placebo-
and metoprolol-treated groups, 19 patients receiving placebo deteriorated to
the point of being listed for cardiac transplantation, a primary end point of
the trial, compared to 2 patients receiving metoprolol. There was significant
improvement in left ventricular ejection fraction, exercise tolerance, NYHA
classification status, and the patients' own assessment of their quality of
life (Waagstein et al., 1993; Andersson et al., 1994). The mean
dose of metoprolol achieved was approximately 100 mg/day following a 6-week
period of gradual upward titration beginning at 10 mg/day. Subsequently, the
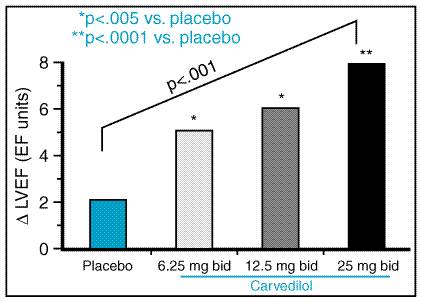
Cardiac Insufficiency Bisoprolol (CIBIS) trial examined the effect of another Carvedilol Carvedilol COREG) is a nonselective
Bisoprolol (ZEBETA CIBIS-II, a follow-up trial to CIBIS I (discussed above), enrolled 2647 patients with moderate to severe heart-failure symptoms due to ischemic or nonischemic dilated cardiomyopathy (CIBIS-II Investigators, 1999). This trial found a 34% reduction in all-cause mortality in bisoprolol-treated patients that was due primarily to a decrease in sudden deaths (44% reduction) and, to a lesser extent, a decrease in pump failure (26% reduction). Because of the way deaths were classified, many patients with pump failure may have been placed in the 'unknown' category, which accounted for a large fraction of all deaths. The mortality benefit of bisoprolol was independent of the etiology of heart failure and was associated with an approximately 36% decrease in hospitalizations for heart failure. Metoprolol (LOPRESSOR, TOPROL XL The Metoprolol Randomized Intervention Trial in Congestive Heart
Failure (MERIT-HF) randomized 3991 patients with NYHA functional class II to
IV symptoms and an ejection fraction <40% to metoprolol (target dose, 200
mg/day) or placebo (MERIT-HF Study Group, 1999). Metoprolol caused a 34%
decrease in all-cause mortality, which was attributable to similar reductions
in sudden death (41% decrease) and death from worsening heart failure (49%
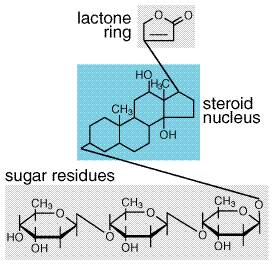
decrease). As in the Mechanism of Action It is not completely clear how Clinical Use of The extensive body of data regarding the use of Since Cardiac Glycosides The cardiac glycosides possess a common molecular motif, a steroid nucleus containing an unsaturated lactone at the C 17 position and one or more glycosidic residues at C 3 (seeFigure 347). Digoxin (LANOXIN, LANOXICAPS) and digitoxin (CRYSTODIGIN) are both orally active, but only digoxin is in widespread clinical use today. Digitoxin differs from digoxin only by the absence of a hydroxyl group at C 12, resulting in a less hydrophilic compound with altered pharmacokinetics compared to digoxin. The cardiac glycosides have been used for centuries as therapeutic agents. The beneficial effects in heart failure were believed to derive from a positive inotropic effect on failing myocardium and efficacy in controlling the ventricular rate response to atrial fibrillation. However, it is now recognized that the cardiac glycosides also modulate sympathetic nervous system activity, an additional mechanism that may contribute importantly to their efficacy in heart failure.
Mechanisms of Action Inhibition of Na+, K+ATPase All cardiac glycosides are potent and highly selective inhibitors of
the active transport of Na+ and K+ across cell
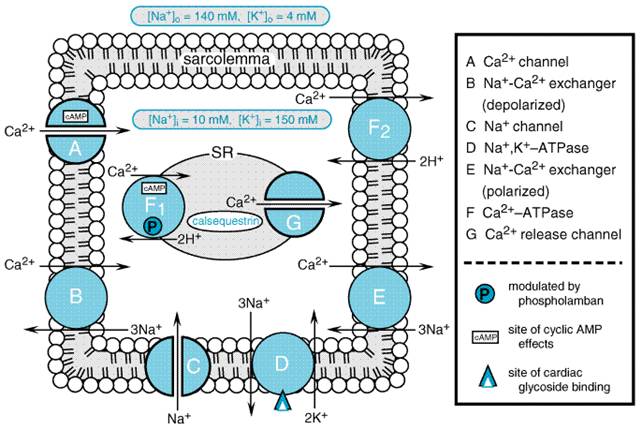
membranes, by binding to a specific site on the extracytoplasmic face of the Positive Inotropic Effect Both Na+ and Ca2+ ions enter cardiac muscle cells during each cycle of depolarization, contraction, and repolarization (Figure 348). Ca2+ that enters the cell via the L-type Ca2+ channel during depolarization triggers the release of additional Ca2+ into the cytosol from an intracellular compartment, the sarcoplasmic reticulum (SR). The greater the amount of activating Ca2+, the greater the force of contraction. During myocyte repolarization and relaxation, Ca2+ is pumped back into the SR by a Ca2+ATPase and also is removed from the cell by the Na+Ca2+ exchanger and by a sarcolemmal Ca2+ATPase.
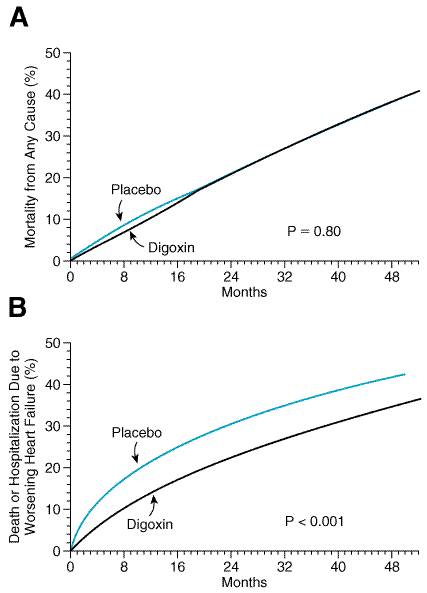
Importantly, the capacity of the exchanger to extrude Ca2+ from the cell depends on the intracellular Na+ concentration. Binding of cardiac glycosides to the sarcolemmal Na+, K+ATPase and inhibition of cellular Na+ pump activity results in a reduction in the rate of active Na+ extrusion and a rise in cytosolic Na+. This increase in intracellular Na+ reduces the transmembrane Na+ gradient driving the extrusion of intracellular Ca2+ during myocyte repolarization. Hence, some incremental Ca2+ is taken up into the SR to be made available to the contractile elements during the subsequent cell depolarization cycle, and contractility of the myocardium is augmented. Electrophysiological Actions (see alsoChapter 35: Antiarrhythmic Drugs) Atrial and ventricular muscle and specialized cardiac pacemaker and conduction fibers exhibit differing responses and sensitivities to cardiac glycosides that are a summation of the direct effects of these drugs on cardiac cells and their indirect, neurally mediated effects. At therapeutic, nontoxic serum or plasma concentrations (i.e., 1.0 to 2.0 ng/ml), digoxin decreases automaticity and increases maximal diastolic resting membrane potential predominantly in atrial and atrioventricular (AV) nodal tissues, due to an increase in vagal tone and a decrease in sympathetic nervous system activity. There also is a prolongation of the effective refractory period and a decrease in conduction velocity in AV nodal tissue. At higher concentrations, this may cause sinus bradycardia or arrest and/or prolongation of AV conduction or heart block. In addition, cardiac glycosides at higher concentrations can increase sympathetic nervous system activity and directly affect automaticity in cardiac tissue, actions that contribute to the generation of atrial and ventricular arrhythmias. Increased intracellular Ca2+ loading and increased sympathetic tone result in an increase in the spontaneous (phase 4) rate of diastolic depolarization as well as delayed afterdepolarizations that may reach the threshold for generation of a propagated action potential. This simultaneous nonuniform increase in automaticity and depression of conduction in HisPurkinje and ventricular muscle fibers predisposes to arrhythmias that may lead to ventricular tachycardia or fibrillation. Regulation of Sympathetic Nervous System Activity An increase in sympathetic nervous system activity is one of the physiological responses to a decline in heart function below that required for maintenance of a cardiac output adequate to meet the metabolic demands of body tissues (i.e., heart failure). This is due, in part, to a reduction in the sensitivity of the arterial baroreflex response to blood pressure, resulting in a decline in tonic baroreflex suppression of CNS-directed sympathetic activity (Ferguson et al., 1989). This desensitization of the normal baroreflex arc also is thought to be responsible in part for the sustained elevation in plasma norepinephrine, renin, and vasopressin levels in heart failure, as well as other indices of systemic neurohumoral activation that are characteristically observed in patients with heart failure. Increased sympathetic nervous system activity initially helps to maintain blood pressure and cardiac output by increasing heart rate, contractility, and systemic vascular resistance, and by decreasing the excretion of salt and water by the kidneys. However, when sustained chronically, these effects of sympathetic overactivity contribute to the pathophysiology of heart failure and progression of the underlying myocardial disease. A direct effect of cardiac glycosides on carotid baroreflex responsiveness to changes in carotid sinus pressure has been demonstrated in isolated baroreceptor preparations from animals with experimental heart failure (Wang et al., 1990). In addition, Ferguson et al. (1989) demonstrated in patients with moderate to advanced heart failure that infusion of the cardiac glycoside deslanoside increased forearm blood flow and cardiac index and decreased heart rate, while markedly decreasing skeletal muscle sympathetic nerve activity, an indicator of the centrally mediated sympathetic nervous system tone. This was unlikely to have been due predominantly to a direct inotropic effect of the drug, since dobutamine, a sympathomimetic drug that increases cardiac output to a comparable extent, did not affect muscle sympathetic nerve activity in these patients. A reduction in neurohumoral activation could represent an important additional mechanism contributing to the efficacy of cardiac glycosides in the treatment of heart failure. Pharmacokinetics The elimination half-life for digoxin is 36 to 48 hours in patients with normal or near-normal renal function. This permits once-a-day dosing for patients with normal or mildly impaired renal function, and near steady-state blood levels are achieved 1 week after initiation of maintenance therapy. Digoxin is excreted for the most part unchanged with a clearance rate that is proportional to the glomerular filtration rate. In patients with congestive heart failure and marginal cardiac reserve, an increase in cardiac output and renal blood flow with vasodilator therapy or sympathomimetic agents may increase renal digoxin clearance, necessitating adjustment of daily maintenance doses. Nevertheless, digoxin is not removed effectively by peritoneal or hemodialysis due to the drug's large (4 to 7 liters/kg) volume of distribution. The principal tissue reservoir is skeletal muscle and not adipose tissue and, thus, dosing should be based on estimated lean body mass. Neonates and infants tolerate and appear to require higher doses of digoxin for an equivalent therapeutic effect than do older children or adults, although absorption and renal clearance rates are similar. Digoxin does cross the placenta, and drug levels in maternal and umbilical vein blood are similar. Most digoxin tablets average 70% to 80% oral bio- availability; however, approximately 10% of the general population harbors the enteric bacterium Eubacterium lentum, which can convert digoxin into inactive metabolites, and this may account for some cases of apparent resistance to standard doses of oral digoxin. Liquid-filled capsules of digoxin (LANOXICAPS) have a higher bioavailability than do tablets and require dosage adjustment if a patient is switched from one dosage form to the other. Parenteral digoxin is available for intravenous administration, and maintenance doses can be given by intravenous injection when oral dosing is impractical. Intramuscular digoxin administration is erratically absorbed, causes local discomfort, and is not recommended. A number of drug interactions (seeTable 344) and clinical conditions can alter digoxin's pharmacokinetics or alter a patient's susceptibility to toxic manifestations of these drugs. Chronic renal failure, for example, decreases digoxin's volume of distribution, necessitating a decrease in maintenance dosage of the drug. Electrolyte disturbances, especially hypokalemia, acid base imbalances, and type of underlying heart disease also may alter a patient's susceptibility to toxic manifestations of digoxin. Clinical Use of Digoxin in Heart Failure Since at least the turn of the century, there has been controversy surrounding the efficacy of cardiac glycosides in the treatment of patients with heart failure who are in sinus rhythm. Despite widespread use of digoxin, objective data from randomized, controlled trials on the safety and efficacy of digoxin had been lacking until the 1990s. The PROVED (Prospective Randomized study Of Ventricular failure and Efficacy of Digoxin; Uretsky et al., 1993) and RADIANCE (Randomized Assessment of Digoxin on Inhibition of Angiotensin Converting Enzyme; Packer et al., 1993) trials examined the effects of withdrawal of digoxin in patients with stable mild to moderate heart failure (i.e., NYHA class II and III) and systolic ventricular dysfunction (left ventricular ejection fraction <0.35). All patients studied were in normal sinus rhythm. Withdrawal of digoxin resulted in a significant worsening of heart-failure symptoms in patients who received placebo compared with patients who continued to receive active drug. Maximal treadmill exercise tolerance also declined significantly in patients withdrawn from digoxin in both trials despite continuation of other medical therapies for heart failure. The much larger Digoxin Investigators' Group (DIG) trial was designed
to detect an effect of digoxin therapy on the survival of patients with heart
failure (The Digitalis Investigation Group, 1997). In this randomized,
double-blind trial, 6,800 patients with predominantly mild to moderate (NYHA
class II to III) heart failure and a left ventricular ejection fraction
<0.45 were assigned to receive either digoxin or placebo in addition to
standard therapy including ACE inhibitors. A trend was seen toward a decrease
in the risk of death attributed to worsening heart failure in the
digoxin-treated group. However, this was balanced by a small increase in the
risk of death due to other cardiac causes (presumed to result from arrhythmia),
and overall, no difference in mortality was seen between the treatment groups
(seeFigure 349). However, fewer patients in the digoxin group were
hospitalized due to worsening heart failure. This benefit was seen at all
levels of ejection fraction but was greatest in patients with more severe
degrees of heart failure. Interestingly, in a predefined substudy of patients
with normal ejection fraction (i.e., presumed to have diastolic
dysfunction), a similar pattern of benefit was seen with digoxin. Based on
these data, it is recommended that digoxin be reserved for patients with
heart failure who are in atrial fibrillation, or for patients in sinus rhythm
who remain symptomatic despite therapy with adequate dosages of ACE
inhibitors and
Doses of Digoxin in Clinical Practice and Monitoring of Serum Levels Using indices of ventricular function, most studies suggest that the greatest increase in contractility is apparent at serum levels of digoxin around 1.4 ng/ml (1.8 nM) (Kelly and Smith, 1992a). The neurohormonal effects of digoxin may occur at lower serum levels, between 0.5 and 1.0 ng/ml; higher serum concentrations than this are not associated with further decreases in neurohormonal activation or with increased clinical benefit. Furthermore, a subgroup analysis of the DIG trial (The Digitalis Investigation Group, 1997) showed an apparent increased risk of death with increasing serum concentrations, even for values within the traditional therapeutic range. Therefore, many authorities advocate maintaining digoxin levels below 1.0 ng/ml. A common approach for initiating digoxin therapy is to begin at 0.125 to 0.25 mg/day, depending on lean body mass and creatinine clearance, and to measure serum digoxin levels a week later when a steady-state has been achieved. The blood sample should be obtained at least 6 hours following the last digoxin dose. Routine surveillance monitoring of digoxin levels need not be carried out, unless a significant deterioration in renal function occurs, or a new drug (e.g., amiodarone) which substantially alters digoxin pharmacokinetics, is started. Oral or intravenous loading with digoxin, while generally safe, is rarely necessary as other safer and more effective drugs exist for short-term inotropic support. Digoxin Toxicity The incidence and severity of digoxin toxicity have declined substantially in the past two decades, due in part to the development of alternative drugs for the treatment of supraventricular arrhythmias and heart failure, to the increased understanding of digoxin pharmacokinetics, to the monitoring of serum digoxin levels, and to the identification of important interactions between digoxin and many commonly used drugs. Nevertheless, the recognition of digoxin toxicity remains an important consideration in the differential diagnosis of arrhythmias and/or neurological and gastrointestinal symptoms in patients receiving cardiac glycosides (Table 345). Vigilance for and early recognition of disturbances of impulse formation, conduction, or both are critically important. Among the more common electrophysiological manifestations are ectopic beats of AV junctional or ventricular origin, first-degree AV block, an excessively slow ventricular rate response to atrial fibrillation, or an accelerated AV junctional pacemaker. These often require only a dosage adjustment and appropriate monitoring. Sinus bradycardia, sinoatrial arrest or exit block, and second- or third-degree AV conduction delay usually respond to atropine, although temporary ventricular pacing may be necessary. Potassium administration should be considered for patients with evidence of increased AV junctional or ventricular automaticity, even when the serum K+ is in the normal range, unless high-grade AV block also is present. Lidocaine or phenytoin, which have minimal effects on AV conduction, may be used for the treatment of worsening ventricular arrhythmias that threaten hemodynamic compromise. Electrical cardioversion carries increased risk of inducing severe rhythm disturbances in patients with overt digitalis toxicity, and it should be used with particular caution. Antidigoxin Immunotherapy An effective antidote for digoxin or digitoxin toxicity is now available in the form of antidigoxin immunotherapy with purified Fab fragments from ovine antidigoxin antisera (DIGIBIND). A full neutralizing dose of Fab based on either the estimated total dose of drug ingested or the total body digoxin burden (Table 346) can be administered intravenously in saline solution over 30 to 60 minutes. For a more comprehensive review of the treatment of digitalis toxicity, seeKelly and Smith (1992b). Chronic Positive Inotropic Therapy Several oral inotropic agents have been developed, some with vasodilating properties. Although many of these agents cause a marked improvement in hemodynamic function and may alleviate symptoms and improve exercise capacity, their effect on mortality with long-term treatment has been disappointing. The dopaminergic agonist ibopamine, the cyclic AMP phosphodiesterase (PDE) inhibitors milrinone, inamrinone (formerly amrinone) and vesnarinone, and the benzimidazoline PDE inhibitor with calcium-sensitizing properties, pimobendan, have been associated with increased mortality (Hampton et al., 1997; Packer et al., 1991; Cohn et al., 1998). This increased mortality has been attributed to an increased risk of arrhythmia with these agents, and it underscores the observation that effects of a drug on hemodynamic function and survival need not be directly related. Therefore, digoxin remains the only oral inotropic agent that should be used in patients with heart failure. Continuous or intermittent outpatient therapy with dobutamine or milrinone, administered by a portable or home-based infusion pump through a central venous catheter, has been evaluated in patients with end-stage heart failure and symptoms refractory to other classes of drugs. There is as yet no convincing evidence that chronic parenteral inotropic therapy improves the quality or length of life. Furthermore, there are concerns that this form of therapy may actually hasten death (reviewed by Gheorghiade, 2000). Outpatient inotropic therapy may be useful in patients with intractable heart failure who are awaiting heart transplantation or who are not candidates for further management in hospital. The use of parenteral inotropic agents in hospitalized patients with heart failure is discussed later in this chapter. Anticoagulation and Antiplatelet Drugs in Heart Failure Patients with heart failure have a significantly higher incidence of stroke and thromboembolism. Nonetheless, the rate of embolic events remains relatively low, and retrospective analyses have shown conflicting evidence of benefit from anticoagulation in patients who remain in sinus rhythm. Anticoagulation with warfarin (COUMADIN; seeChapter 55: Anticoagulant, Thrombolytic, and Antiplatelet Drugs) is recommended for patients who have atrial fibrillation or a history of a previous embolic event, if there is evidence of a left ventricular thrombus, or possibly, if left ventricular dysfunction is severe and the ventricles are markedly dilated. Similarly, while aspirin therapy is effective in reducing the incidence of ischemic events in patients with coronary artery disease, concerns have been raised that aspirin may attenuate the benefits from ACE inhibitors in patients with heart failure (Nguyen et al., 1997; Al-Khadra et al., 1998). A potential mechanism for this postulated adverse interaction is antagonism of bradykinin-mediated prostaglandin generation by aspirin. Synthesis of vasodilatory prostaglandins and other potentially beneficial molecules is reduced, thereby attenuating the favorable effects of ACE inhibition. In support of this thesis is the finding that the vasodilatory effects of enalapril may be blunted by concomitant administration of aspirin (Hall et al., 1992). Furthermore, a recent study has shown a trend toward greater morbidity and mortality in patients with heart failure receiving aspirin compared to those receiving placebo or warfarin (Cleland, 1999). Accordingly, until more definitive data are available on the use of aspirin in heart failure, it should be reserved for patients who have a clear indication for its use (e.g., the presence of known or suspected coronary artery disease). Alternative antiplatelet agents that do not appear to interfere with prostaglandin metabolism are now available in the form of the adenosine diphosphate antagonists ticlopidine (TICLID) and clopidogrel (PLAVIX). While both agents have been demonstrated to be effective in the prevention of ischemic events, there are as yet few data on their use in patients who have heart failure. Antiarrhythmic Drugs in Heart Failure Sudden cardiac death accounts for a significant proportion of the mortality due to heart failure. In the majority of cases, these deaths have been attributed to a ventricular tachyarrhythmia. However, ventricular arrhythmias are common in patients in heart failure, and are usually asymptomatic. Antiarrhythmic drugs, while effective in suppressing ventricular arrhythmias, may be associated with an increased risk of mortality, believed related to the proarrhythmic and negative inotropic properties common to many of these agents (Echt et al., 1991) (seeChapter 35: Antiarrhythmic Drugs). Therefore, indiscriminate use of antiarrhythmic drugs is not recommended in patients with heart failure who have ventricular arrhythmias. A possible exception is amiodarone (CORDARONE; seeChapter 35:
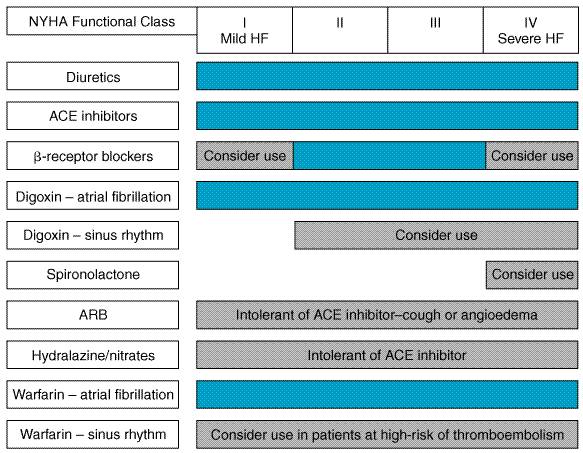
Antiarrhythmic Drugs), an antiarrhythmic agent that has additional Thus, the available evidence does not support the routine use of amiodarone to suppress ventricular arrhythmias in patients with heart failure. Patients who have experienced a life-threatening ventricular arrhythmia should receive an ICD as treatment of first choice, with amiodarone being reserved as alternative therapy for patients who are unable to receive an ICD. However, amiodarone is effective in preventing the recurrence of atrial fibrillation or other supraventricular arrhythmias in patients who have ventricular dysfunction. Guidelines for the Treatment of Ambulatory Heart Failure As the number of proven therapeutic interventions in heart failure grows, the complexity of managing these patients has correspondingly increased. A number of guidelines for the management of patients with heart failure have been published by learned societies. The main points of the most recent set of guidelines (Heart Failure Society of America, 1999) are summarized in Figure 3410.
|
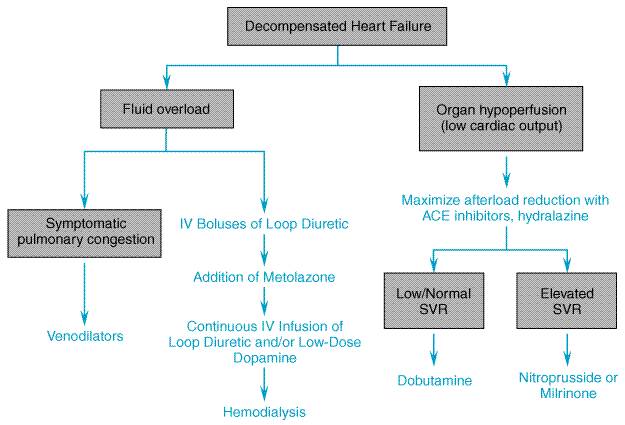
Parenteral Drugs for the Treatment of Hospitalized Patients with Heart Failure
|
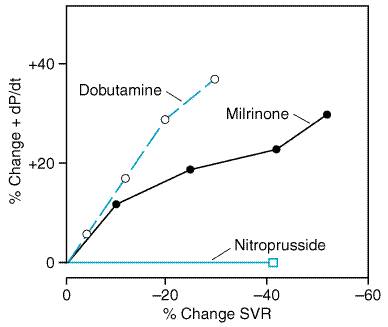
General Considerations Patients with heart failure are commonly hospitalized because of increased dyspnea and peripheral edema due to pulmonary and systemic congestion. Fatigue also is a frequent complaint and is related to a reduction in cardiac output and perfusion of skeletal muscle. Accordingly, relief of congestion through the use of diuretics and possibly venodilators is often a priority in patients hospitalized for heart failure. Treatment also is directed at improving ventricular function and increasing cardiac output by lowering ventricular afterload and enhancing myocardial contractility. Wherever possible, precipitating factors (e.g., fever, infection) should be identified and removed, and the underlying cause of heart failure (e.g., ischemia, valvular disease) corrected. Diuretics As discussed in the treatment of ambulatory heart failure, diuretics
are important for the alleviation of intravascular and extravascular fluid
overload. In patients with decompensated heart failure of sufficient severity
to warrant admission to a hospital, it is generally desirable to initiate an
effective diuresis by using intravenous doses of a loop diuretic.
Intravenous administration provides a more rapid and predictable diuresis
than does the oral route, which is susceptible to delayed or impaired gut
absorption, particularly in patients with marked fluid accumulation. The loop
diuretic may be administered as repetitive boluses which are titrated against
the desired response, or by constant infusion. An advantage of the latter
approach is that the same total daily dose of diuretic, when given as a
continuous infusion, can result in a more sustained and continuous
natriuresis due to maintenance of high diuretic drug levels within the lumen
of renal tubules. In addition, the constant infusion helps to decrease the
risk of ototoxicity that occurs with transient, high blood levels of the drug
following repetitive intermittent loop diuretic dosing (Lahav et al.,
1992). A typical continuous furosemide infusion is initiated with a
40-mg bolus injection followed by a constant infusion of 10 mg/hour, with
upward titration of the infusion as necessary. When there is a poor response
to diuretics due to reduced renal perfusion, the short-term administration of
sympathomimetic drugs or phosphodiesterase inhibitors to increase cardiac
output may be necessary to achieve a response. Another useful approach (see
section on Diuretic Resistance in Heart Failure) is the intravenous
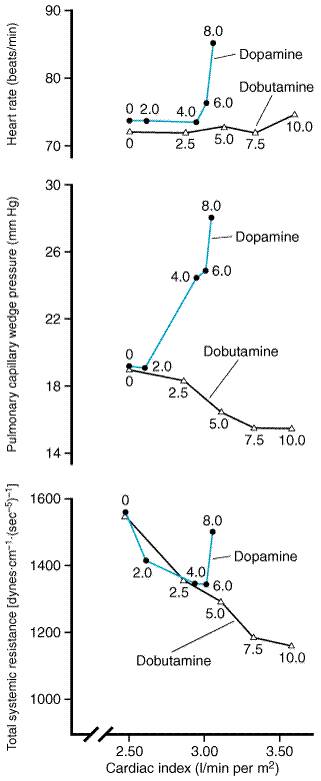
administration of dopamine at so-called 'low' doses (i.e.,
less than 2 Parenteral Vasodilators Sodium Nitroprusside Sodium nitroprusside NITROPRESS) is a potent vasodilator that is effective in reducing both ventricular filling pressures and systemic and arterial resistance. It has a rapid onset of action, within 2 to 5 minutes, is quickly metabolized to cyanide and nitric oxide, and its dose usually can be titrated expeditiously to achieve an optimal and predictable hemodynamic effect. For these reasons, nitroprusside is commonly used in intensive-care settings for rapid control of hypertension (seeChapter 33: Antihypertensive Agents and the Drug Therapy of Hypertension) and for the management of acutely decompensated heart failure. Nitroprusside reduces ventricular filling pressures by directly increasing venous compliance, resulting in a redistribution of blood volume from central to peripheral veins. Nitroprusside is among the most effective afterload-reducing drugs by virtue of the spectrum of pharmacodynamic actions the drug has on different vascular beds (seeFigure 3411). It causes a fall in peripheral vascular resistance as well as an increase in aortic wall compliance and, at optimal doses, improves ventricularvascular coupling. These effects decrease left-ventricular afterload, resulting in an increase in cardiac output. Nitroprusside also dilates pulmonary arterioles and reduces right ventricular afterload. This combination of preload- and afterload-reducing effects improves myocardial energetics by reducing wall stress, provided that blood pressure does not fall to the point of compromising diastolic coronary artery flow or of activating a marked reflex increase in sympathetic nervous system tone. Following the rapid withdrawal of nitroprusside infusion, there may be a transient deterioration in ventricular function associated with a 'rebound' increase in systemic vascular resistance that is thought to reflect activation of neurohormonal systems. Nitroprusside is particularly effective in patients with congestive heart failure due to mitral regurgitation or left-to-right shunts through a ventricular septal defect.
The most common adverse effect of nitroprusside, as with most vasodilators, is hypotension. The increase in renal blood flow that accompanies an increase in cardiac output following initiation of nitroprusside in patients with severe heart failure may improve glomerular filtration and diuretic effectiveness. However, the redistribution of blood flow from central organs to peripheral vascular beds may limit or prevent an increase in renal blood flow in some patients. Cyanide produced during the biotransformation of nitroprusside is rapidly metabolized by the liver to thiocyanate, which is cleared by the kidney. Thiocyanate and/or cyanide toxicity is uncommon but may occur in the face of hepatic or renal failure, or following prolonged high-dose infusions of nitroprusside. Typical symptoms include unexplained abdominal pain, mental status changes, convulsions or lactic acidosis. Methemoglobinemia is another unusual complication of prolonged, high-dose nitroprusside infusion. Nitroglycerin Nitroglycerin, like nitroprusside, is a potent vasodilator (see previous section on the treatment of ambulatory heart failure and Chapter 32: Drugs Used for the Treatment of Myocardial Ischemia). In contrast to nitroprusside, nitroglycerin is relatively selective for veins, particularly at low infusion rates. Thus, intravenous nitroglycerin is most often used in the treatment of acute heart failure when a decrease in ventricular filling pressures is desired. At higher infusion rates, nitroglycerin also causes decreases in the systemic and pulmonary arterial resistance, thereby decreasing ventricular afterload. The use of nitroglycerin may be limited by headache and the development of nitrate tolerance, although the latter is generally overcome by uptitration of the infusion rate to the desired response. Since nitroglycerin is administered in ethanol, high infusion rates can be associated with significant blood-alcohol levels.
Dopamine and dobutamine are the positive inotropic agents most often used for the short-term support of the circulation in advanced heart failure. Isoproterenol, epinephrine, and norepinephrine, although useful in specific circumstances, have little role in the treatment of most cases of severe heart failure. The basic pharmacology of these and other adrenergic agonists is discussed in Chapter 10: Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists. Dobutamine Dobutamine DOBUTREX), in the formulation available
for clinical use, is a racemic mixture that stimulates both
Continuous
infusions of dobutamine for up to several days generally are well tolerated,
although pharmacological tolerance may limit the efficacy during long-term
use. Infusion rates usually are initiated at 2 to 3 Dopamine Dopamine,
an endogenous catecholamine, has limited utility in the treatment of most
patients with systolic ventricular dysfunction who are not in shock due to
primary cardiac failure, hemorrhage, dehydration, or toxicity from
vasodilatory drugs. The pharmacological and hemodynamic effects of dopamine
are strongly dose- related. At low doses of less than or equal to 2 At intermediate infusion rates (2 to 5 Phosphodiesterase Inhibitors The cyclic AMP PDE inhibitors mimic the effects of adenylyl cyclase activation by inhibiting the breakdown of cyclic AMP. Increased levels of cyclic AMP result in increased myocardial contractility and vasodilation in the venous and arterial circulation. Although agents such as theophylline and caffeine had been identified as nonspecific cyclic GMP and cyclic AMP PDE inhibitors more than 30 years ago, the use of these drugs at hemodynamically effective doses is plagued by side effects. During the 1980s, amrinone (now called inamrinone), milrinone, and other PDE inhibitors with isoenzyme selectivity became available, largely alleviating these problems. Inamrinone and Milrinone Parenteral formulations of inamrinone (INOCOR) and milrinone (PRIMACOR) have been approved for short-term support of the circulation in advanced heart failure. Both drugs are bipyridine derivatives and relatively selective inhibitors of the cyclic GMP-inhibited, cyclic AMP PDE (type III) family. These drugs cause direct stimulation of myocardial contractility and acceleration of myocardial relaxation. In addition, they cause balanced arterial and venous dilation with a consequent fall in systemic and pulmonary vascular resistances, and left and right heart filling pressures. Cardiac output increases due to the stimulation of myocardial contractility and the decrease in left ventricular afterload. As a result of this dual mechanism of action, the increase in cardiac output with milrinone, when compared at comparable decreases in systemic pressure, is greater than with the pure vasodilator nitroprusside; and conversely, the arterial and venous dilator effects of milrinone are greater, when compared for comparable increases in cardiac output, than with dobutamine (Colucci et al., 1986) (seeFigure 3411). Both drugs are effective when given either as single agents or, more
commonly, in combination with other oral and/or intravenous drugs for
short-term treatment of patients with severe heart failure due to systolic
right or left ventricular dysfunction. Intravenous infusions of either drug
should be initiated by a loading dose followed by a continuous infusion. For inamrinone,
typically a 0.75-mg/kg bolus injection over 2 to 3 minutes is followed by a
2- to 20- General Guidelines for the Use of Parenteral Drugs In general, initial therapy in the patient with symptomatic pulmonary congestion should include diuresis to alleviate pulmonary and systemic vascular congestion (seeFigure 3413). The administration of an oral or intravenous nitrate preparation may provide additional rapid symptomatic relief of pulmonary congestion by causing venodilation. Cardiac output may be optimized by a variety of agents, the selection of which may be guided in part by the arterial pressure, or ideally, by knowledge of the systemic vascular resistance (SVR) as provided by a pulmonary artery catheter.
In patients with an elevated arterial pressure, afterload reduction with nitroprusside may be very effective. Nitroprusside also may be effective when there is an elevated SVR, despite the presence of a low arterial pressure. On the other hand, in patients in whom the SVR is not elevated, afterload reduction may result in an excessive fall in arterial pressure. In such patients, increasing myocardial contractility with a positive inotropic agent such as dobutamine often is preferable. Milrinone, which exerts both inotropic and vasodilatory actions, may be tolerated in some patients who do not tolerate a pure vasodilator, and conversely, may provide better relief of pulmonary congestion than dobutamine. Likewise, in patients in whom the left ventricular filling pressure is known (or suspected) not to be markedly elevated, treatment with dobutamine may avoid an excessive decrease in preload that can occur with vasodilators. When heart failure is severe, in patients who fail to respond to standard therapy, or if there is progressive renal insufficiency in the face of persistent signs of elevated filling pressures, placement of a pulmonary artery catheter and hemodynamic monitoring may be important for guidance of drug selection and dosing. Once hemodynamic monitoring is established, therapy is directed at optimization of hemodynamics and improvement in clinical status of the patient; afterload and cardiac filling pressures should be reduced to target levels, and cardiac output augmented where necessary to ensure adequate perfusion of vital organs. |
Prospectus
|
Since the publication of the previous edition
of this textbook, there has been continuing evolution in the drug treatment
of heart failure. Several large randomized trials have shown consistent
evidence of benefit and have firmly established the central role of Despite these and other recent advances in the pharmacotherapy of
heart failure, the mortality rate remains high. Thus, preventive strategies
including aggressive control of risk factors for cardiovascular disease
continue to be of great importance. Further insights into the cellular and
molecular abnormalities that underlie the contractile, hemodynamic, and
neurohormonal disturbances of heart failure have cleared the way for the
development of innovative therapeutic agents that may impact positively on
outcomes. New therapeutic strategies include agents that block or stimulate
endogenous signaling pathways. Endothelin receptor blockers have been
shown to exert beneficial hemodynamic effects in patients with heart failure
during short-term administration and are being evaluated with regard to
long-term effects on morbidity and mortality. Antagonists of the
inflammatory cytokine, tumor necrosis factor- For further discussion of normal and abnormal cardiac function and of
heart failure seeChapters 215 and 216 in Harrison's Principles of
Internal Medicine, 16th ed., Acknowledgment The authors wish to acknowledge Drs. Ralph A. Kelley and Thomas W. Smith, the authors of this chapter in the ninth edition of Goodman and Gilman's The Pharmacological Basis of Therapeutics, some of whose text has been retained in this edition. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2853
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved