| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Principles of Toxicology and Treatment of Poisoning
Overview
|
Chemicals that are developed into drugs must have therapeutic efficacy and be safe. Unfortunately, all chemicals have the potential to produce unwanted effects. Therefore, in the development of drugs, it is essential to select chemicals that have a margin of safety between the dose that produces the desired (therapeutic) effect and the dose that produces undesired (toxic) effects. The margin of safety for some drugs is small, and some people intentionally overdose themselves. As a result, toxic effects of drugs often are observed. The physician must be aware that the symptoms a patient manifests might be due to chemical exposure, either to a drug or another chemical. This chapter summarizes the principles of how chemicals produce toxic effects as well as the principles for the treatment of poisoning. |
Principles of Toxicology
|
Toxicology is the science of the adverse effects of chemicals on living organisms. The discipline often is divided into several major areas. The descriptive toxicologist performs toxicity tests (described below) to obtain information that can be used to evaluate the risk that exposure to a chemical poses to human beings and to the environment. The mechanistic toxicologist attempts to determine how chemicals exert deleterious effects on living organisms. Such studies are essential for the development of tests for the prediction of risks, for facilitating the search for safer chemicals, and for rational treatment of the manifestations of toxicity. The regulatory toxicologist judges whether or not a drug or other chemical has a low enough risk to justify making it available for its intended purpose. The Food and Drug Administration (FDA) regulates drugs, medical devices, cosmetics, and food additives in interstate commerce. For food additives, the FDA attempts to determine the acceptable daily intake (ADI) that can be consumed over an entire lifetime without any appreciable risk. The Environmental Protection Agency (EPA) is responsible for regulation of pesticides, toxic chemicals, hazardous wastes, and toxic pollutants in water and air. The Occupational Safety and Health Administration (OSHA) determines whether or not employers are providing working conditions that are safe for employees. Employers must keep the concentration of each chemical in the air of the workplace below a threshold limit value (TLV). The Consumer Products Safety Commission regulates all articles sold for use in homes, in schools, or for recreation except those products regulated by the FDA and the EPA. Two specialized areas of toxicology are particularly important for medicine. Forensic toxicology, which combines analytical chemistry and fundamental toxicology, is concerned with the medicolegal aspects of chemicals. Forensic toxicologists assist in postmortem investigations to establish the cause or circumstances of death. Clinical toxicology focuses on diseases that are caused by or are uniquely associated with toxic substances. Clinical toxicologists treat patients who are poisoned by drugs and other chemicals and develop new techniques for the diagnosis and treatment of such intoxications. The physician must evaluate the possibility that a patient's signs and symptoms might be caused by toxic chemicals present in the environment or administered as therapeutic agents. Many of the adverse effects of drugs mimic symptoms of disease. Appreciation of the principles of toxicology is necessary for the recognition and management of such clinical problems. |
DoseResponse Relationship
|
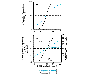
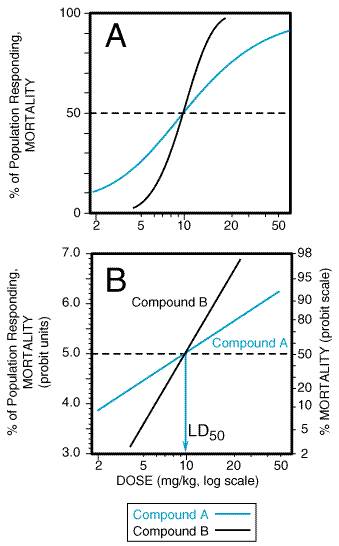
Evaluation of the doseresponse or the doseeffect relationship is crucially important to toxicologists. There is a graded doseresponse relationship in an individual and a quantal doseresponse relationship in the population (see Chapter 3: Principles of Therapeutics). Graded doses of a drug given to an individual usually result in a greater magnitude of response as the dose is increased. In a quantal doseresponse relationship, the percentage of the population affected increases as the dose is raised; the relationship is quantal in that the effect is specified to be either present or absent in a given individual (see Figure 33). This quantal doseresponse phenomenon is extremely important in toxicology and is used to determine the median lethal dose (LD50) of drugs and other chemicals. The LD50 is determined experimentally. The chemical being evaluated usually is administered to mice or rats (orally or intraperitoneally) at several doses (usually four or five) in the lethal range (Figure 41A). To linearize such data, the response (death) can be converted to units of deviation from the mean, or probits (from the contraction of probability units). The probit designates the deviation from the median; a probit of 5 corresponds to a 50% response, and, because each probit equals one standard deviation, a probit of 4 equals 16% and a probit of 6 equals 84%. A plot of percent of population responding, in probit units, against log dose yields a straight line (Figure 41B). The LD50 is determined by drawing a vertical line from the point on the line where the probit unit equals 5 (50% mortality). The slope of the doseeffect curve also is important. The LD50 for both compounds depicted in Figure 41 is the same (10 mg/kg). However, the slopes of the doseresponse curves are quite different. At a dose equal to one half of the LD50 (5 mg/kg), less than 5% of the animals exposed to compound B would die, but 30% of the animals given compound A would die.
The quantal, or 'all-or-none,' response is not limited to lethality. As described in Chapter 3: Principles of Therapeutics, similar doseeffect curves can be constructed for any effect produced by chemicals. |
Risk and Its Assessment
|
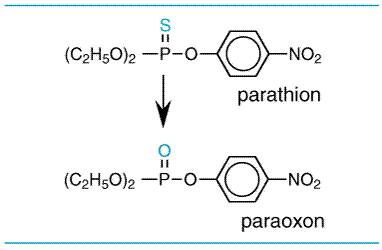
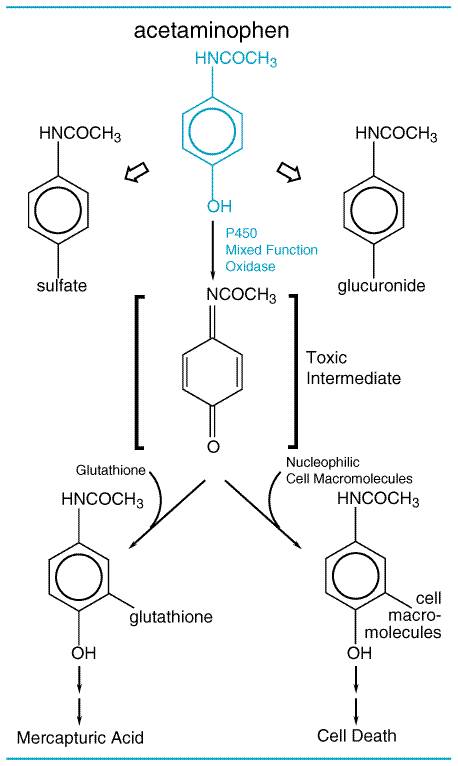
There are marked differences in the LD50's of various chemicals. Some result in death at doses of a fraction of a microgram (LD50 for botulinum toxin equals 10 pg/kg); others may be relatively harmless in doses of several grams or more. While categories of toxicity that are of some practicality have been devised, based on the amount required to produce death, often it is not easy to distinguish between toxic and nontoxic chemicals. Paracelsus (14931541) noted that 'All substances are poisons; there is none which is not a poison. The right dose differentiates a poison and a remedy.' Although society wants the toxicologist to categorize all chemicals as either safe or toxic, this is not possible. The real concern is the risk associated with use of the chemical, not whether or not a chemical is toxic. In the assessment of risk, one also must consider the harmful effects of the chemical accrued directly or indirectly through adverse effects on the environment when used in the quantity and in the manner proposed. Depending on the use and disposition of a chemical, a very toxic compound ultimately may be less harmful than a relatively nontoxic one. At present there is much concern about the risk from exposure to chemicals that have produced cancer in laboratory animals. For most of these chemicals, it is not known if they also produce cancer in human beings. The regulatory agencies take one of three approaches to potential chemical carcinogens. For food additives, the FDA is very cautious, as large numbers of people are likely to be exposed to the chemicals, and they are not likely to have beneficial effects to individuals. For drugs, the FDA weighs the relative risks and benefits of the drugs for patients. Thus, it is unlikely that the FDA will approve the use of a drug that produces tumors in laboratory animals for a mild ailment, but it may approve its use for a serious disease. In fact, most cancer chemotherapeutic drugs also are chemical carcinogens. In the regulation of environmental carcinogens, the EPA attempts to limit lifetime exposure such that the incidence of cancer due to the chemical would be no more than one in a million people. To determine the daily allowable exposure for human beings, mathematical models are used to extrapolate doses of chemicals that produce a particular incidence of tumors in laboratory animals (often in the range of 10% to 20%) to those that should produce cancer in no more than one person in a million. The models used are conservative and are thought to provide adequate protection from undue risks from exposure to potential carcinogens. Acute versus Chronic Exposure Effects of acute exposure to a chemical often differ from those that follow subacute or chronic exposure. Acute exposure occurs when a dose is delivered as a single event. Chronic exposure is likely to be to small quantities of a substance over a long period of time, which often results in the slow accumulation of the compound in the body. Evaluation of cumulative toxic effects is receiving increased attention because of chronic exposure to low concentrations of various natural and synthetic chemical substances in the environment. Chemical Forms of Drugs That Produce Toxicity The 'parent' drug administered to the patient often is the chemical form producing the desired therapeutic effect. Similarly, the toxic effects of drugs often are due to deleterious effects of the parent drug. However, toxic effects of drugs (as well as therapeutic effects) and other chemicals also can be due to metabolites of the drug produced by enzymes, light, or reactive oxygen species. Toxic Metabolites The metabolites of many chemicals are responsible for their toxicities. Most organophosphate insecticides are biotransformed by the cytochrome P450 system to produce their toxicities; for example, parathion is biotransformed to paraoxon (seeFigure 42). Paraoxon is a stable metabolite that binds to and inactivates cholinesterase. Some metabolites of drugs are not chemically stable and are referred to as reactive intermediates. An example of a toxic reactive intermediate is the metabolite of acetaminophen (seeFigure 43), which is very reactive and binds to nucleophiles such as glutathione; when cellular glutathione is depleted, the metabolite binds to cellular macromolecules, the mechanism by which acetaminophen kills liver cells. Both parathion and acetaminophen are more toxic under conditions in which the cytochrome P450 enzymes are increased, such as following ethanol or phenobarbital exposure, because these are responsible for the production of the toxic metabolites (seeChapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination).
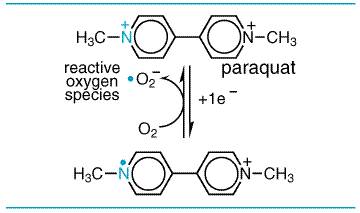
Phototoxic and Photoallergic Reactions Many chemicals are activated to toxic metabolites by hepatic enzymatic biotransformation. However, some chemicals can be activated in the skin by ultraviolet and/or visible radiation. In photoallergy, radiation absorbed by a drug, such as a sulfonamide, results in its conversion to a product that is a more potent allergen than the parent compound. Phototoxic reactions to drugs, in contrast to photoallergic ones, do not have an immunological component. Drugs, either absorbed locally into the skin or that have reached the skin through the systemic circulation, may be the object of photochemical reactions within the skin; this can lead directly either to chemically induced photosensitivity reactions or to enhancement of the usual effects of sunlight. Tetracyclines, sulfonamides, chlorpromazine, and nalidixic acid are examples of phototoxic chemicals; generally, they are innocuous to skin if not exposed to light. Reactive Oxygen Species Paraquat is an herbicide that produces severe lung injury. Its toxicity is not due to paraquat or its metabolites but rather to reactive oxygen species formed during one-electron reduction of paraquat paired with electron donation to oxygen (seeFigure 44).
|
Spectrum of Undesired Effects
|
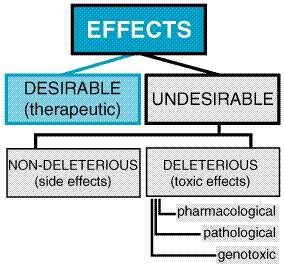
The spectrum of undesired effects of chemicals may be broad and ill-defined (see Figure 45). In therapeutics, a drug typically produces numerous effects, but usually only one is sought as the primary goal of treatment; most of the other effects are referred to as undesirable effects of that drug for that therapeutic indication. Side effects of drugs usually are nondeleterious; they include effects such as dry mouth occurring with tricyclic antidepressant therapy. Mechanistic categorization of toxic effects is a necessary prelude to their avoidance or, if they occur, to their rational and successful management.
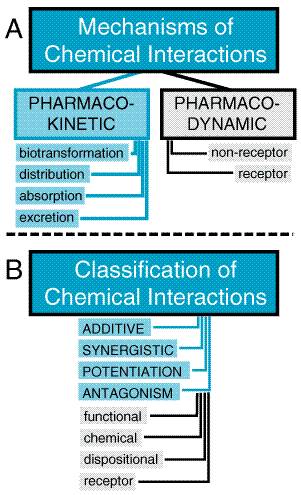
Types of Toxic Reactions Toxic effects of drugs may be classified as pharmacological, pathological, or genotoxic (alterations of DNA), and their incidence and seriousness are related, at least over some range, to the concentration of the toxic chemical in the body. An example of a pharmacological toxicity is excessive depression of the central nervous system (CNS) by barbiturates; an example of a pathological effect is hepatic injury produced by acetaminophen; an example of a genotoxic effect is a neoplasm produced by a nitrogen mustard. If the concentration of chemical in the tissues does not exceed a critical level, the effects usually will be reversible. The pharmacological effects usually disappear when the concentration of drug or chemical in the tissues is decreased by biotransformation or excretion from the body. Pathological and genotoxic effects may be repaired. If these effects are severe, death may ensue within a short time; if more subtle damage to DNA is not repaired, cancer may appear in a few months or years in laboratory animals or in a decade or more in human beings. Local versus Systemic Toxicity Local toxicity is the effect that occurs at the site of first contact between the biological system and the toxicant. Local effects can be caused by ingestion of caustic substances or inhalation of irritant materials. Systemic toxicity requires absorption and distribution of the toxicant; most substances, with the exception of highly reactive chemical species, produce systemic toxic effects. The two categories are not mutually exclusive. Tetraethyllead, for example, injures skin at the site of contact and deleteriously affects the CNS after it is absorbed into the circulation. Most systemic toxicants affect one or a few organs predominantly. The target organ of toxicity is not necessarily the site of accumulation of the chemical. For example, lead is concentrated in bone, but its primary toxic action is on soft tissues; DDT (chlorophenothane) is concentrated in adipose tissue but produces no known toxic effects there. The CNS is involved in systemic toxicity most frequently, as many compounds with prominent effects elsewhere also affect the brain. Next in order of frequency of involvement in systemic toxicity are the circulatory system; the blood and hematopoietic system; visceral organs such as liver, kidney, and lung; and the skin. Muscle and bone are least often affected. With substances that have a predominantly local effect, the frequency of tissue reaction depends largely on the portal of entry (skin, gastrointestinal tract, or respiratory tract). Reversible and Irreversible Toxic Effects The effects of drugs on human beings must, whenever possible, be reversible; otherwise, the drugs would be prohibitively toxic. If a chemical produces injury to a tissue, the capacity of the tissue to regenerate or recover will largely determine the reversibility of the effect. Injuries to a tissue such as liver, which has a high capacity to regenerate, usually are reversible; injury to the CNS is largely irreversible, because the highly differentiated neurons of the brain have an extremely limited ability to divide and regenerate. Delayed Toxicity Most toxic effects of drugs occur at a predictable (usually short) time after administration. However, such is not always the case. For example, aplastic anemia caused by chloramphenicol may appear weeks after the drug has been discontinued. Carcinogenic effects of chemicals usually have a long latency period, and 20 to 30 years may pass before tumors are observed. Because such delayed effects cannot be assessed during any reasonable period of initial evaluation of a chemical, there is an urgent need for reliably predictive, short-term tests for such toxicity as well as for systematic surveillance of the long-term effects of marketed drugs and other chemicals (see Chapter 3: Principles of Therapeutics). Chemical Carcinogens Chemical carcinogens are classified into two major groups, genotoxic and nongenotoxic. Genotoxic carcinogens interact with DNA, whereas nongenotoxic carcinogens do not. Chemical carcinogenesis is a multistep process. Most genotoxic carcinogens are themselves unreactive (procarcinogens or proximate carcinogens) but are converted to primary or ultimate carcinogens in the body. The drug-metabolizing enzymes (phase I and phase II) often convert the proximate carcinogens to reactive electron-deficient intermediates (electrophiles). These reactive intermediates can interact with electron-rich (nucleophilic) centers in DNA to produce a mutation. Such interaction of the ultimate carcinogen with DNA in a cell is thought to be the initial step in chemical carcinogenesis. The DNA may revert to normal if DNA repair mechanisms operate successfully; if not, the transformed cell may grow into a tumor that becomes apparent clinically. Nongenotoxic carcinogens, also referred to as promoters, do not produce tumors alone but do potentiate the effects of genotoxic carcinogens. Promotion involves facilitation of the growth and development of so-called dormant or latent tumor cells. The time from initiation to the development of a tumor probably depends on the presence of such promoters; for many human tumors, the latent period is 15 to 45 years. To determine whether or not a chemical is potentially carcinogenic to humans, two main types of laboratory tests are done. One type of study is performed to determine whether or not the chemical is mutagenic, because many carcinogens are also mutagens. These studies are often in vitro studies, such as the Ames test using Salmonella typhimurium (Ames et al., 1975), which can be completed within a few days. This type of test can detect genotoxic carcinogens but not promoters. The second type of study to detect chemical carcinogens consists of feeding laboratory animals (mice and rats) the chemical at high dosages for their entire life span. Autopsies and histopathological examinations are performed on each animal. The incidence of tumors in control animals and animals fed the chemical are compared to determine whether the chemical produces an increased incidence of tumors. This latter study can detect promoters as well as genotoxic carcinogens. Allergic Reactions Chemical allergy is an adverse reaction that results from previous sensitization to a particular chemical or to one that is structurally similar. Such reactions are mediated by the immune system. The terms hypersensitivity and drug allergy often are used to describe the allergic state. For a low-molecular-weight chemical to cause an allergic reaction, it or its metabolic product usually acts as a hapten, combining with an endogenous protein to form an antigenic complex. Such antigens induce the synthesis of antibodies, usually after a latent period of at least 1 or 2 weeks. Subsequent exposure of the organism to the chemical results in an antigenantibody interaction that provokes the typical manifestations of allergy. Doseresponse relationships usually are not apparent for the provocation of allergic reactions. Allergic responses have been divided into four general categories, based on the mechanism of immunological involvement (Coombs and Gell, 1975). Type I, or anaphylactic, reactions in human beings are mediated by IgE antibodies. The Fc portion of IgE can bind to receptors on mast cells and basophils. If the Fab portion of the antibody molecule then binds antigen, various mediators (histamine, leukotrienes, prostaglandins) are released and cause vasodilation, edema, and an inflammatory response. The main targets of this type of reaction are the gastrointestinal tract (food allergies), the skin (urticaria and atopic dermatitis), the respiratory system (rhinitis and asthma), and the vasculature (anaphylactic shock). These responses tend to occur quickly after challenge with an antigen to which the individual has been sensitized and are termed immediate hypersensitivity reactions. Type II, or cytolytic, reactions are mediated by both IgG and IgM antibodies and usually are attributed to their ability to activate the complement system. The major target tissues for cytolytic reactions are the cells in the circulatory system. Examples of type II allergic responses include penicillin-induced hemolytic anemia, methyldopa-induced autoimmune hemolytic anemia, quinidine-induced thrombocytopenic purpura, and sulfonamide-induced granulocytopenia. Fortunately, these autoimmune reactions to drugs usually subside within several months after removal of the offending agent. Type III, or Arthus, reactions are mediated predominantly by IgG; the mechanism involves the generation of antigenantibody complexes that subsequently fix complement. The complexes are deposited in the vascular endothelium, where a destructive inflammatory response called serum sickness occurs. This phenomenon contrasts with the type II reaction, in which the inflammatory response is induced by antibodies directed against tissue antigens. The clinical symptoms of serum sickness include urticarial skin eruptions, arthralgia or arthritis, lymphadenopathy, and fever. These reactions usually last for 6 to 12 days and then subside after the offending agent is eliminated. Several drugssuch as sulfonamides, penicillins, certain anticonvulsants, and iodidescan induce serum sickness. StevensJohnson syndrome, such as that caused by sulfonamides, is a more severe form of immune vasculitis. Symptoms of this reaction include erythema multiforme, arthritis, nephritis, CNS abnormalities, and myocarditis. Type IV, or delayed-hypersensitivity, reactions are mediated by sensitized T lymphocytes and macrophages. When sensitized cells come in contact with antigen, an inflammatory reaction is generated by the production of lymphokines and the subsequent influx of neutrophils and macrophages. An example of type IV or delayed hypersensitivity is the contact dermatitis caused by poison ivy. Idiosyncratic Reactions Idiosyncrasy is defined as a genetically determined abnormal reactivity to a chemical. The observed response is qualitatively similar in all individuals, but the idiosyncratic response may take the form of extreme sensitivity to low doses or extreme insensitivity to high doses of chemicals. These genetic polymorphisms can be due to interindividual differences in drug pharmacokinetics, such as phase I and phase II biotransformation enzymes. An example is increased incidence of peripheral neuropathy in patients with inherited deficiencies in acetylation when isoniazid is used for the treatment of tuberculosis. The polymorphisms also can be due to pharmacodynamic factors such as drug-receptor interactions (Evans and Relling, 1999). For example, many black males (about 10%) develop a serious hemolytic anemia when they receive primaquine as an antimalarial therapy. Such individuals have a deficiency of erythrocytic glucose-6-phosphate dehydrogenase (see Chapter 40: Drugs Used in the Chemotherapy of Protozoal Infections: Malaria). Genetically determined resistance to the anticoagulant action of warfarin is due to an alteration in the vitamin K epoxide reductase (see Chapter 55: Anticoagulant, Thrombolytic, and Antiplatelet Drugs). The use of genetic information to explain interindividual differences in drug responses or to individualize dosages of drugs for patients with known genetic polymorphisms is referred to as pharmacogenomics. Interactions between Chemicals The existence of numerous toxicants requires consideration of their potential interactions (see Figure 46). Concurrent exposures may alter the pharmacokinetics of drugs by changing rates of absorption, the degree of protein binding, or the rates of biotransformation or excretion of one or both interacting compounds. The pharmacodynamics of chemicals can be altered by competition at the receptor; for example, atropine is used to treat organophosphate insecticide toxicity, because it blocks muscarinic cholinergic receptors and prevents their stimulation by excess acetylcholine resulting from inhibition of acetylcholinesterase by the insecticide. Nonreceptor pharmacodynamic drug interactions also can occur when two drugs have different mechanisms of action; for example, aspirin and heparin given together can cause unexpected bleeding. The response to combined toxicants may thus be equal to, greater than, or less than the sum of the effects of the individual agents.
Numerous terms describe pharmacological and toxicological interactions (see Figure 46B). An additive effect describes the combined effect of two chemicals that is equal to the sum of the effect of each agent given alone; the additive effect is the most common. A synergistic effect is one in which the combined effect of two chemicals is greater than the sum of the effects of each agent given alone. For example, both carbon tetrachloride and ethanol are hepatotoxins, but together they produce much more injury to the liver than expected from the mathematical sum of their individual effects. Potentiation is the increased effect of a toxic agent acting simultaneously with a nontoxic one. Isopropanol alone, for example, is not hepatotoxic; however, it greatly increases the hepatotoxicity of carbon tetrachloride. Antagonism is the interference of one chemical with the action of another. An antagonistic agent is often desirable as an antidote. Functional or physiological antagonism occurs when two chemicals produce opposite effects on the same physiological function. For example, this principle is applied to the ability of an intravenous infusion of dopamine to maintain perfusion of vital organs during certain severe intoxications characterized by marked hypotension. Chemical antagonism or inactivation is a reaction between two chemicals to neutralize their effects. For example, dimercaprol, or British antilewisite (BAL), chelates with various metals to decrease their toxicity (see Chapter 67: Heavy Metals and Heavy-Metal Antagonists). Dispositional antagonism is the alteration of the disposition of a substance (its absorption, biotransformation, distribution, or excretion) so that less of the agent reaches the target organ or its persistence there is reduced (see below). Antagonism at the receptor for the chemical entails the blockade of the effect of an agonist with an appropriate antagonist that competes for the same site. For example, the antagonist naloxone is used to treat respiratory depression produced by opioids (see Chapter 23: Opioid Analgesics). |
Descriptive Toxicity Tests in Animals
|
Two main principles underlie all descriptive toxicity tests performed in animals. First, effects of chemicals produced in laboratory animals, when properly qualified, apply to toxicity in human beings. When calculated on the basis of dose per unit of body surface, toxic effects in human beings usually are encountered in the same range of concentrations as are those in experimental animals. On the basis of body weight, human beings are generally more vulnerable than experimental animals. Such information is used to select dosages for clinical trials of candidate therapeutic agents and to attempt to set limits on permissible exposure to environmental toxicants. The second main principle is that exposure of experimental animals to toxic agents in high doses is a necessary and valid method to discover possible hazards to human beings who are exposed to much lower doses. This principle is based on the quantal doseresponse concept. As a matter of practicality, the number of animals used in experiments on toxic materials usually will be small compared with the size of human populations potentially at risk. For example, 0.01% incidence of a serious toxic effect (such as cancer) represents 25,000 people in a population of 250 million. Such an incidence is unacceptably high. Yet, detecting an incidence of 0.01% experimentally would probably require a minimum of 30,000 animals. To estimate risk at low dosage, large doses must be given to relatively small groups. The validity of the necessary extrapolation is clearly a crucial question. Chemicals are first tested for toxicity by estimation of the LD50 in two animal species by two routes of administration; one of these is the expected route of exposure of human beings to the chemical being tested. The number of animals that die in a 14-day period after a single dose is recorded. The animals also are examined for signs of intoxication, lethargy, behavioral modification, and morbidity. The chemical is next tested for toxicity by subacute exposure, usually for 90 days. The subacute study is performed most often in two species by the route of intended use or exposure, and at least three doses are employed. A variety of parameters are monitored during this period, and, at the end of the study, organs and tissues are examined by a pathologist. Long-term or chronic studies are carried out in animals at the same time that clinical trials are undertaken (see Chapter 3: Principles of Therapeutics). For drugs, the length of exposure depends somewhat on the intended clinical use. If the drug normally would be used for short periods under medical supervision, as would an antimicrobial agent, a chronic exposure of animals for 6 months might suffice. If the drug would be used in human beings for longer periods, a study of chronic use for 2 years might be required. Studies of chronic exposure often are used to determine the carcinogenic potential of chemicals. These studies usually are performed in rats and mice for the average lifetime of the species. Other tests are designed to evaluate teratogenicity (congenital malformations), perinatal and postnatal toxicity, and effects on fertility. Teratogenicity studies usually are performed by administering drugs to pregnant rats and rabbits during the period of organogenesis. In addition to chronic studies for evaluation of carcinogenic
potential or teratogenicity, drugs often are tested for mutagenic
potential. The most popular such test currently available, the reverse
mutation test developed by |
Incidence of Acute Poisoning
|
The true incidence of poisoning in the Deaths in the The substances most frequently involved in human poison exposures are shown in Table 41. Two of the three categories of substances most frequently responsible for human poisoning are not drugs but cosmetics and cleaning agents. While most drugs are not the most common class of chemicals involved in human poisoning, the top five categories of substances that produce deaths are drugs (Table 42). The chemicals most commonly associated with fatalities are tricyclic antidepressants, acetaminophen, salicylates, opiates, cocaine, digoxin, carbon monoxide, and calcium channel blockers. Most of the people who die from poisoning are adults, and the deaths often result from intentional rather than accidental exposure. Children younger than 6 years of age account for 53% of the poisoning incidents reported but only 2% of the deaths. Children between 1 and 2 years of age have the highest incidence of accidental poisoning. Fortunately, most of the substances available to these young children are not highly toxic. Iron and pesticides are the leading cause of pediatric accidental poisoning fatalities. It recently has been recognized that the incidence of serious and
fatal adverse drug reactions in |
Major Sources of Information on Poisoning
|
Pharmacology textbooks are a good source of information on treatment of poisoning by drugs, but they usually say little about other chemicals. Additional information on drugs and other chemicals can be found in various books on poisoning. (See Ellenhorn, 1997; Goldfrank et al., 1998; Haddad et al., 1998; Klaassen, 2001.) A useful source of information on the treatment of acute poisoning by
commercial products is Clinical Toxicology of Commercial Products by
Gosselin and associates (1984). This book contains seven major sections. One
section lists more than 17,500 trade names of products that might be ingested
accidentally or suicidally. It lists the manufacturer and ingredients of each
commercial product and notes components believed responsible for harmful
effects. A popular computerized system for information on toxic substances is
POISINDEX (Micromedex, Inc., There are about 120 poison control centers in the |
Prevention and Treatment of Poisoning
|
Many acute poisonings from drugs could be prevented if physicians provided common-sense instructions about the storage of drugs and other chemicals and if patients or parents of patients accepted this advice. These instructions are so widely publicized that they need not be repeated here. For clinical purposes, all toxic agents can be divided into two classes: those for which a specific treatment antidote exists and those for which there is no specific treatment. For the vast majority of drugs and other chemicals, there is no specific treatment; symptomatic medical care that supports vital functions is the only strategy. Supportive therapy, as in other medical emergencies, is the most important aspect of the treatment of drug poisoning. The adage 'Treat the patient, not the poison' remains the most basic and important principle of clinical toxicology. Maintenance of respiration and circulation takes precedence. Serial measurement and charting of vital signs and important reflexes help to judge the progress of intoxication, response to therapy, and need for additional treatment. This monitoring usually requires hospitalization. The classification in Table 43 often is used to indicate the severity of CNS intoxication. Treatment with large doses of stimulants and sedatives often can cause more harm than the poison. Chemical antidotes should be used judiciously; heroic measures seldom are necessary. Treatment of acute poisoning must be prompt. The first goal is to maintain the vital functions if their impairment is imminent. The second goal is to keep the concentration of poison in the crucial tissues as low as possible by preventing absorption and enhancing elimination. The third goal is to combat the pharmacological and toxicological effects at the effector sites. Prevention of Further Absorption of Poison Emesis Although emesis is indicated after poisoning by oral ingestion of most chemicals, it is contraindicated in certain situations: (1) If the patient has ingested a corrosive poison, such as a strong acid or alkali (e.g., drain cleaners), emesis increases the likelihood of gastric perforation and further necrosis of the esophagus. (2) If the patient is comatose or in a state of stupor or delirium, emesis may cause aspiration of the gastric contents. (3) If the patient has ingested a CNS stimulant, further stimulation associated with vomiting may precipitate convulsions. (4) If the patient has ingested a petroleum distillate (e.g., kerosene, gasoline, or petroleum-based liquid furniture polish), regurgitated hydrocarbons can be aspirated readily and cause chemical pneumonitis (Ervin, 1983). In contrast, emesis should be considered if the solution that is ingested contains potentially dangerous compounds, such as pesticides. There are marked differences in the capabilities of various petroleum distillates to produce hydrocarbon pneumonia, which is an acute, hemorrhagic necrotizing process. In general, the ability of various hydrocarbons to produce pneumonitis is inversely proportional to the viscosity of the agent: if the viscosity is high, as with oils and greases, the risk is limited; if the viscosity is low, as with mineral seal oil found in liquid furniture polishes, the risk of aspiration is high. Vomiting can be induced mechanically by stroking the posterior pharynx. However, this technique is not as effective as the administration of ipecac or apomorphine. Ipecac The most useful household emetic is syrup of ipecac (not ipecac fluid extract, which is 14 times more potent and may cause fatalities). Syrup of ipecac is available in 0.5- and 1-fluid ounce containers (approximately 15 and 30 ml), which may be purchased without prescription. The drug can be given orally, but it takes 15 to 30 minutes to produce emesis; this compares favorably with the time usually required for adequate gastric lavage. The oral dose is 15 ml in children from 6 months to 12 years of age and 30 ml in older children and adults. Because emesis may not occur when the stomach is empty, the administration of ipecac should be followed by a drink of water. Ipecac acts as an emetic because of its local irritant effect on the enteric tract and its effect on the chemoreceptor trigger zone (CTZ) in the area postrema of the medulla. Syrup of ipecac may be effective even when antiemetic drugs (such as phenothiazines) have been ingested (Thoman and Verhulst, 1966), presumably due to its direct irritant action on the gastrointestinal tract. Ipecac can produce toxic effects on the heart because of its content of emetine, but this usually is not a problem with the dose used for emesis (Manno and Manno, 1977). If emesis does not occur, ipecac should be removed by gastric lavage. Chronic abuse of ipecac for weight reduction can result in cardiomyopathy, ventricular fibrillation, and death. Apomorphine Apomorphine stimulates the CTZ and causes emesis. The drug is unstable in solution and must be prepared just prior to use and thus is not often readily available. Additionally, apomorphine is not effective orally and must be given parenterally, usually by the subcutaneous route6 mg for adults and 0.06 mg/kg for children (Goldfrank et al., 1998). However, this can be an advantage over ipecac in that it can be administered to an uncooperative patient and produces vomiting in 3 to 5 minutes. Because apomorphine is a respiratory depressant, it should not be used if the patient has been poisoned by a CNS depressant or if the patient's respiration is slow and labored. At present, apomorphine is rarely used as an emetic. Gastric Lavage Gastric lavage is accomplished by inserting a tube into the stomach
and washing the stomach with water, normal saline, or one-half normal saline
to remove the unabsorbed poison. The procedure should be performed as soon as
possible, but only if vital functions are adequate or supportive procedures
have been implemented. The contraindications to this procedure generally are
the same as for emesis, and there is the additional potential complication of
mechanical injury to the throat, esophagus, and stomach. A position statement
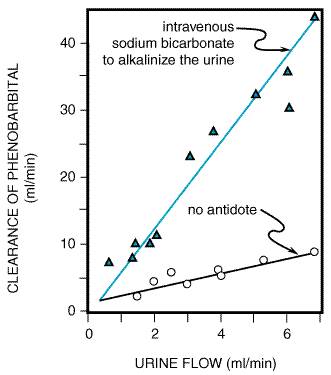
has been published by the The only equipment needed for gastric lavage is a tube and a large syringe. The tube should be as large as possible so that the wash solution, food, and poison (whether in the form of a capsule, pill, or liquid) will flow freely and lavage can be accomplished quickly. A 36-Fr tube or larger should be used in adults and a 24-Fr tube or larger in children. Orogastric lavage is preferred over nasogastric, because a larger tube can be employed. To prevent aspiration, an endotracheal tube with an inflatable cuff should be positioned before lavage is initiated if the patient is comatose, having seizures, or has lost the gag reflex. During gastric lavage, the patient should be placed on his or her left side because of the anatomical asymmetry of the stomach, with the head hanging face down over the edge of the examining table. If possible, the foot of the table should be elevated. This technique minimizes chances of aspiration. The contents of the stomach should be aspirated with an irrigating syringe and saved for chemical analysis. The stomach then may be washed with saline solution. Saline solution is safer than water in young children because of the risk of water intoxication, manifested by tonic and clonic seizures and coma (Arena, 1975). Only small volumes (120 to 300 ml) of lavage solution should be instilled into the stomach at one time so that the poison is not pushed into the intestine. Lavage should be repeated until the returns are clear, which usually requires 10 to 12 washings and a total of 1.5 to 4 liters of lavage fluid. When the lavage is complete, the stomach may be left empty or an antidote may be instilled through the tube. If no specific antidote is known for the poison, an aqueous suspension of activated charcoal and a cathartic is often given. Chemical Adsorption Activated charcoal avidly adsorbs drugs and chemicals on the surfaces of the charcoal particles, thereby preventing absorption and toxicity. Many, but not all, chemicals are adsorbed by charcoal. For example, alcohols, hydrocarbons, metals, and corrosives are not well adsorbed by activated charcoal, and charcoal therefore is of little value in treating these poisonings. The effectiveness of charcoal also is dependent on the time since the ingestion and on the dose of charcoal; one should attempt to achieve a charcoal:drug ratio of at least 10:1. Activated charcoal also can interrupt the enterohepatic circulation of drugs and enhance the net rate of diffusion of the chemical from the body into the gastrointestinal tract. For example, the use of serial doses of activated charcoal has been shown to enhance the elimination of theophylline and phenobarbital (Berg et al., 1982; Berlinger et al., 1983). During the last two decades, there has been an increase in the use of activated charcoal and a corresponding decrease in the use of ipecac-induced emesis and gastric lavage in the treatment of poisoning. Studies in patients with drug overdoses as well as in normal subjects fail to show a benefit of treatment with ipecac or lavage plus activated charcoal as compared with charcoal alone (Neuvonen et al., 1983; Curtis et al., 1984; Kulig et al., 1985; Albertson et al., 1989). It is concluded generally that adminstration of activated charcoal is the single most important intervention that can be provided to an overdosed patient. Activated charcoal usually is prepared as a mixture of at least 50 g (about 10 heaping tablespoons) in a glass of water. The mixture is then administered either orally or via a gastric tube. Because most poisons do not appear to desorb from the charcoal if charcoal is present in excess, the adsorbed poison need not be removed from the gastrointestinal tract. Activated charcoal should not be used simultaneously with ipecac because charcoal can adsorb the emetic agent in ipecac and thus reduce the drug's emetic effect. Charcoal also may adsorb and decrease the effectiveness of specific antidotes. Activated charcoal must be distinguished from the so-called universal antidote, which consists of two parts burned toast (not activated charcoal), one part tannic acid (strong tea), and one part magnesium oxide. In practice, the universal antidote is ineffective. As mentioned, the presence of an adsorbent in the intestine may interrupt enterohepatic circulation of a toxicant, thus enhancing its excretion. Activated charcoal is useful in interrupting the enterohepatic circulation of drugs such as tricyclic antidepressants and glutethimide. A nonabsorbable polythiol resin has been used to treat poisoning by methylmercury due to its ability to bind mercury excreted into the bile (see Chapter 67: Heavy Metals and Heavy-Metal Antagonists). Cholestyramine hastens the elimination of cardiac glycosides by a similar mechanism. Chemical Inactivation Antidotes can change the chemical nature of a poison by rendering it less toxic or preventing its absorption. Formaldehyde poisoning can be treated with ammonia to form hexamethylenetetramine (Goldstein et al., 1974); sodium formaldehyde sulfoxylate can convert mercuric ion to the less soluble metallic mercury (Gosselin et al., 1984); and sodium bicarbonate converts ferrous iron to ferrous carbonate, which is poorly absorbed. Chemical inactivation techniques seldom are used today, however, because valuable time may be lost, whereas emetics, activated charcoal, and gastric lavage are rapid and effective. In the past, neutralization was the usual treatment of poisoning with acids or bases. Vinegar, orange juice, or lemon juice often has been used for the patient who has ingested alkali, and various antacids often have been advocated for treatment of acid burns. The use of neutralizing agents is controversial, because it may produce excessive heat. Carbon dioxide gas produced from bicarbonates used to treat oral poisoning with acids can cause gastric distention and even perforation. The treatment of choice for ingestion of either acids or alkalis is dilution with water or milk. Similarly, burns produced by acid or alkali on the skin should be treated with copious amounts of water. Purgation The rationale for using an osmotic cathartic is to minimize absorption by hastening the passage of the toxicant through the gastrointestinal tract. Few, if any, controlled clinical data are available on the effectiveness of cathartics in the treatment of poisoning. Cathartics generally are considered harmless unless the poison has injured the gastrointestinal tract. Cathartics are indicated after the ingestion of enteric-coated tablets, when the time after ingestion is greater than 1 hour, and for poisoning by volatile hydrocarbons (Rumack and Lovejoy, 1985). Sorbitol is the most effective, but sodium sulfate and magnesium sulfate also are used; all act promptly and usually have minimal toxicity. However, magnesium sulfate should be used cautiously in patients with renal failure or in those likely to develop renal dysfunction, and Na+-containing cathartics should be avoided in patients with congestive heart failure. Whole-bowel irrigation is a technique that not only promotes defecation, but also eliminates the entire contents of the intestines. This technique uses a high-molecular-weight polyethylene glycol and isosmolar electrolyte solution (PEG-EESS), which does not alter serum electrolytes. It is commercially available as GOLYTELY and COLYTE Inhalation and Dermal Exposure to Poisons When a poison has been inhaled, the first priority is to remove the patient from the source of exposure. Similarly, the skin should be thoroughly washed with water if it has come in contact with a poison. Contaminated clothing should be removed. Initial treatment of all types of chemical injuries to the eye must be rapid; thorough irrigation of the eye with water for 15 minutes should be performed immediately. Enhanced Elimination of the Poison Biotransformation Once a chemical has been absorbed, procedures sometimes can be employed to enhance its rate of elimination. Many drugs are metabolized by the cytochrome P450 system in the endoplasmic reticulum of the liver, and components of this system can be induced by a number of compounds (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination). However, induction of these oxidative enzymes is too slow (days) to be valuable in the treatment of acute poisoning by most chemical agents. Many chemicals are toxic because they are biotransformed into more toxic chemicals. Thus, inhibition of biotransformation should decrease the toxicity of such drugs. For example, ethanol is used to inhibit the conversion of methanol to its highly toxic metabolite, formic acid, by alcohol dehydrogenase (see Chapter 68: Nonmetallic Environmental Toxicants: Air Pollutants, Solvents and Vapors, and Pesticides). As discussed earlier in this chapter, acetaminophen is converted by the cytochrome P450 system to an electrophilic metabolite that is detoxified by glutathione, a cellular nucleophile. Acetaminophen does not cause hepatotoxicity until glutathione is depleted, whereupon the reactive metabolite binds to essential macromolecular constituents of the hepatocyte, resulting in cell death. The liver can be protected by maintenance of the concentration of glutathione, and this can be accomplished by the administration of N-acetylcysteine (Black, 1980; see Chapter 27: Analgesic-Antipyretic and Antiinflammatory Agents and Drugs Employed in the Treatment of Gout). Some drugs are detoxified by conjugation with glucuronic acid or sulfate before elimination from the body, and the availability of the endogenous cosubstrates for conjugation may limit the rate of elimination; such is the case in the detoxication of acetaminophen (Hjelle et al., 1985). When methods become available to replete these compounds, an additional mechanism will be available to treat poisoning. Similarly, detoxication of cyanide by conversion to thiocyanate can be accelerated by the administration of thiosulfate (see Chapter 68: Nonmetallic Environmental Toxicants: Air Pollutants, Solvents and Vapors, and Pesticides). Biliary Excretion The liver excretes many drugs and other foreign chemicals into bile, but little is known about efficient ways to enhance biliary excretion of xenobiotics for the treatment of acute poisoning. Inducers of microsomal enzyme activity enhance biliary excretion of some xenobiotics, but the effect is slow in onset (Klaassen and Watkins, 1984). Urinary Excretion Drugs and poisons are excreted into the urine by glomerular filtration and active tubular secretion (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination); they can be reabsorbed into the blood if they are in a lipid-soluble form that will penetrate the tubule or if there is an active mechanism for their transport. There are no methods known to accelerate the active transport of poisons into urine, and enhancement of glomerular filtration is not a practical means to facilitate elimination of toxicants. However, passive reabsorption from the tubular lumen can be altered. Diuretics decrease reabsorption by decreasing the concentration gradient of the drug from the lumen to the tubular cell and by increasing flow through the tubule. Furosemide is used most often, but osmotic diuretics also are employed (see Chapter 29: Diuretics). Forced diuresis should be used with caution, especially in patients with renal, cardiac, or pulmonary complications. Nonionized compounds are reabsorbed far more rapidly than ionized, polar molecules; therefore a shift from the nonionized to the ionized species of the toxicant by alteration of the pH of the tubular fluid may hasten elimination (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination). Acidic compounds such as phenobarbital and salicylates are cleared much more rapidly in alkaline than in acidic urine. The effect of increasing urine flow and alkalinization of urine on the clearance of phenobarbital is shown in Figure 47. Intravenous sodium bicarbonate is used to alkalinize the urine. Renal excretion of basic drugs such as amphetamine theoretically can be enhanced by acidification of the urine. Acidification can be accomplished by the administration of ammonium chloride or ascorbic acid. Urinary excretion of an acidic compound is particularly sensitive to changes in urinary pH if its pKa is within the range of 3.0 to 7.5; for bases the corresponding range is 7.5 to 10.5.
Dialysis Hemodialysis or hemoperfusion usually has limited use in the treatment of intoxication with chemicals. However, under certain circumstances, such procedures can be lifesaving. The utility of dialysis depends on the amount of poison in the blood relative to the total body burden. Thus, if a poison has a large volume of distribution, as is the case for the tricyclic antidepressants, the plasma will contain too little of the compound for effective removal by dialysis. Extensive binding of the compound to plasma proteins impairs dialysis greatly. The kinetics of elimination of a toxicant by dialysis also is dependent on the rate of dissociation of the compound from binding sites in tissues; for some chemicals, this rate may be slow. Although peritoneal dialysis requires a minimum of personnel and can be started as soon as the patient is admitted to the hospital, it is too inefficient to be of value for the treatment of acute intoxications. Hemodialysis (extracorporeal dialysis) is much more effective than peritoneal dialysis and may be essential in a few life-threatening intoxications, such as with methanol, ethylene glycol, and salicylates. Passage of blood through a column of charcoal or adsorbent resin
(hemoperfusion) is a technique for the extracorporeal removal of a poison ( Antagonism or Chemical Inactivation of an Absorbed Poison The functional and pharmacological antagonism of the effects of absorbed toxicants has been discussed above. If a patient is poisoned with a compound that acts as an agonist at a receptor for which a specific blocking agent is available, administration of the receptor antagonist may be highly effective. Functional antagonism also can be valuable for support of the patient's vital functions. For example, anticonvulsant drugs are used to treat chemically induced convulsions. However, drugs that stimulate antagonistic physiological mechanisms are not always of clinical value and may even decrease the incidence of survival, because it often is difficult to titrate the effect of one drug against another when the two act on opposing systems. An example of such a complication is the use of CNS stimulants to attempt to reverse respiratory depression. Convulsions are a typical complication of such therapy, and mechanical support of respiration is preferred. In addition, the duration of action of the poison and the antidote may differ, sometimes leading to poisoning with the antidote. Specific chemical antagonists of a toxicant, such as opioid antagonists (see Chapter 23: Opioid Analgesics) and atropine as an antagonist of pesticide-induced acetylcholine excess (Chapter 7: Muscarinic Receptor Agonists and Antagonists), are valuable but unfortunately rare. Chelating agents with high selectivity for certain metal ions provide such examples (see Chapter 67: Heavy Metals and Heavy-Metal Antagonists). Antibodies offer the potential for the production of specific antidotes for a host of common poisons and for drugs that frequently are abused or misused. A notable example of such success is the use of purified digoxin-specific Fab fragments of antibodies in the treatment of potentially fatal cases of poisoning with digoxin (see Chapter 34: Pharmacological Treatment of Heart Failure). The development of human monoclonal antibodies directed against specific toxins has significant potential therapeutic value. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3279
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved