| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antimicrobial Agents: Drugs Used in the Chemotherapy of Tuberculosis, Mycobacterium avium Complex Disease, and Leprosy
Overview
|
The pharmacological characteristics and the
therapeutic use of each class of compounds employed in the chemotherapy of
tuberculosis, diseases caused by the Mycobacterium avium complex, and
leprosy are discussed in this chapter. The increase in tuberculosis case
rates in the |
Antimicrobial Agents: Drugs Used in the Chemotherapy of Tuberculosis, Mycobacterium Avium Complex Disease, and Leprosy: Introduction
|
Drugs used in the treatment of tuberculosis can be divided into two major categories (seeTable 481). 'First-line' agents combine the greatest level of efficacy with an acceptable degree of toxicity; these include isoniazid, rifampin, ethambutol, streptomycin, and pyrazinamide. The large majority of patients with tuberculosis can be treated successfully with these drugs. Excellent results for patients with nondrug-resistant tuberculosis can be obtained with a 6-month course of treatment; for the first 2 months, isoniazid, rifampin, and pyrazinamide are given, followed by isoniazid and rifampin for the remaining 4 months. Administration of rifampin in combination with isoniazid for 9 months also is effective therapy for all forms of disease caused by strains of Mycobacterium tuberculosis susceptible to both agents (Bass et al., 1994). In areas where primary resistance to isoniazid occurs, therapy usually is initiated with four drugsrifampin, isoniazid, pyrazinamide, and ethambutol (or streptomycin)until sensitivity tests are completed. Occasionally, however, because of microbial resistance, it may be necessary to resort to 'second-line' drugs in addition, so that treatment may be initiated with 5 to 6 drugs. This category of agents includes ofloxacin, ciprofloxacin, ethionamide, aminosalicylic acid, cycloserine, amikacin, kanamycin, and capreomycin (seeIseman, 1993). In HIV-infected patients receiving protease inhibitors and/or nonnucleoside reverse transcriptase inhibitors, drug interactions with the rifamycins (rifampin, rifapentine, rifabutin) are an important concern. Directly observed therapy improves the outcome of tuberculosis treatment regimens (Havlir and Barnes, 1999). Isoniazid is ineffective in the treatment of leprosy or M. avium complex infection. Lepromatous (multibacillary) leprosy is treated with dapsone, clofazimine, and rifampin for a minimum of 2 years, while tuberculoid (paucibacillary) leprosy is treated with dapsone and rifampin for 6 months. Antimicrobial agents with activity against M. avium complex include rifabutin, clarithromycin, azithromycin, and fluoroquinolones. Clarithromycin and azithromycin are more effective than rifabutin for prophylaxis of M. avium complex infection in patients with AIDS. Clarithromycin or azithromycin, in combination with ethambutol (to prevent development of resistance), is effective treatment for M. avium complex infection in HIV-infected individuals. |
Drugs for Tuberculosis
|
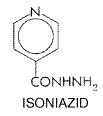
Isoniazid Isoniazid (isonicotinic acid hydrazide; NYDRAZID, others) is still considered to be the primary drug for the chemotherapy of tuberculosis, and all patients with disease caused by isoniazid-sensitive strains of the tubercle bacillus should receive the drug if they can tolerate it. History The discovery of isoniazid was somewhat fortuitous. In 1945, Chorine reported that nicotinamide possesses tuberculostatic action. Examination of the compounds related to nicotinamide revealed that many pyridine derivatives possess tuberculostatic activity; among these are congeners of isonicotinic acid. Because the thiosemicarbazones were known to inhibit M. tuberculosis, the thiosemicarbazone of isonicotinaldehyde was synthesized and studied. The starting material for this synthesis was the methyl ester of isonicotinic acid, and the first intermediate was isonicotinylhydrazide (isoniazid). The interesting history of these chemical studies has been reviewed by Fox (1953). Chemistry Isoniazid is the hydrazide of isonicotinic acid; the structural formula is as follows:
The isopropyl derivative of isoniazid, iproniazid (1-isonicotinyl-2-isopropylhydrazide), also inhibits the multiplication of the tubercle bacillus. This compound, which is a potent inhibitor of monoamine oxidase, is too toxic for use in human beings. However, its study led to the use of monoamine oxidase inhibitors for the treatment of depression (seeChapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). Antibacterial Activity Isoniazid is bacteriostatic for 'resting' bacilli but is
bactericidal for rapidly dividing microorganisms. The minimal tuberculostatic
concentration is 0.025 to 0.05 Isoniazid is highly effective for the treatment of experimentally induced tuberculosis in animals and is strikingly superior to streptomycin. Unlike streptomycin, isoniazid penetrates cells with ease and is just as effective against bacilli growing within cells as it is against those growing in culture media. Among the various nontuberculous (atypical) mycobacteria, only M. kansasii is usually susceptible to isoniazid. However, sensitivity always must be tested in vitro, since the inhibitory concentration required may be rather high. Bacterial Resistance When tubercle bacilli are grown in vitro in increasing concentrations of isoniazid, mutants are readily selected that are resistant to the drug, even when the drug is present in enormous concentrations. However, cross-resistance between isoniazid and other agents used to treat tuberculosis (except ethionamide, which is structurally related to isoniazid) does not occur. The most common mechanism of isoniazid resistance is mutations in catalase-peroxidase that decrease its activity, preventing conversion of the prodrug isoniazid to its active metabolite (Blanchard, 1996). Another mechanism of resistance is related to a missense mutation within the mycobacterial inhA gene involved in mycolic acid biosynthesis (Banerjee et al., 1994). As with the other agents described, treatment with isoniazid alone
leads to the emergence in vivo of resistant strains. The shift from
primarily sensitive to mainly insensitive microorganisms occasionally occurs
within a few weeks after therapy is started; however, the time of appearance
of this phenomenon varies considerably from one case to another.
Approximately one in 106 tubercle bacilli will be genetically
resistant to isoniazid; since tuberculous cavities may contain as many as 107
to 109 microorganisms, it is not surprising that treatment with
isoniazid alone results in the selection of these resistant bacteria. The
incidence of primary resistance to isoniazid in the Mechanism of Action Takayama and associates (1975) were among the first to suggest that a
primary action of isoniazid is to inhibit the biosynthesis of mycolic acids,
important constituents of the mycobacterial cell wall. The mechanism of
action of isoniazid is complex, with resistance mapping to mutations in at
least five different genes (katG, inhA, ahpC, kasA,
and ndh). The preponderance of evidence points to inhA as the
primary drug target. The inhA gene encodes the enoyl-ACP reductase of
fatty acid synthase II, which converts Absorption, Distribution, and Excretion Isoniazid is readily absorbed when administered either orally or
parenterally. Aluminum-containing antacids may interfere with absorption. Peak
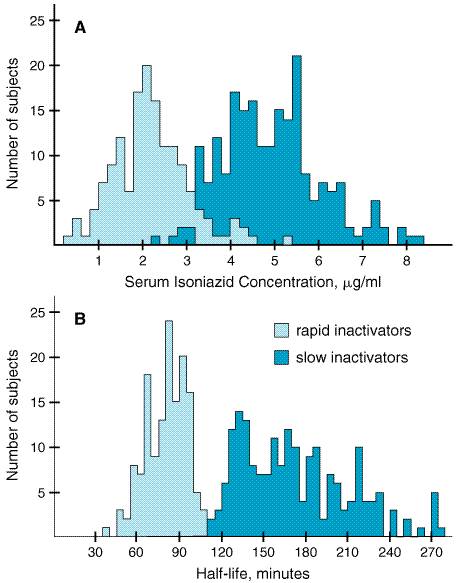
plasma concentrations of 3 to 5 Isoniazid diffuses readily into all body fluids and cells. The drug is detectable in significant quantities in pleural and ascitic fluids; concentrations in the cerebrospinal fluid (CSF) with inflamed meninges are similar to those in the plasma (Holdiness, 1985). Isoniazid penetrates well into caseous material. The concentration of the agent is initially higher in the plasma and muscle than in the infected tissue, but the latter retains the drug for a long time in quantities well above those required for bacteriostasis. From 75% to 95% of a dose of isoniazid is excreted in the urine within 24 hours, mostly as metabolites. The main excretory products in human beings are the result of enzymatic acetylation (acetylisoniazid) and enzymatic hydrolysis (isonicotinic acid). Small quantities of an isonicotinic acid conjugate (probably isonicotinyl glycine), one or more isonicotinyl hydrazones, and traces of N-methylisoniazid also are detectable in the urine. Human populations show genetic heterogeneity with regard to the rate of acetylation of isoniazid (Evans et al., 1960). The distribution of slow and rapid inactivators of the drug is bimodal owing to differences in the activity of an acetyltransferase (Figure 481). The rate of acetylation significantly alters the concentrations of the drug that are achieved in plasma and its half-life in the circulation. The half-life of the drug may be prolonged in the presence of hepatic insufficiency.
The
frequency of each acetylation phenotype is dependent upon race but is not
influenced by gender or age. Fast acetylation is found in Inuit and Japanese.
Slow acetylation is the predominant phenotype in most Scandinavians, Jews,
and North African Caucasians. The incidence of 'slow acetylators'
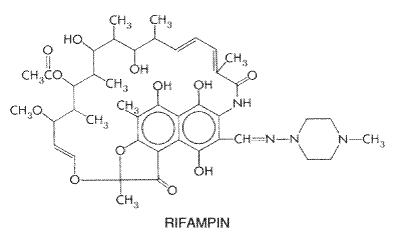
among the various racial types in the The clearance of isoniazid is dependent to only a small degree on the status of renal function, but patients who are slow inactivators of the drug may accumulate toxic concentrations if their renal function is impaired. Bowersox and colleagues (1973) have suggested that 300 mg per day of the drug can be administered safely to individuals in whom the plasma creatinine concentration is less than 12 mg/dl (1.1 mM). Therapeutic Uses Isoniazid is still the most important drug worldwide for the treatment of all types of tuberculosis. Toxic effects can be minimized by prophylactic therapy with pyridoxine and careful surveillance of the patient. The drug must be used concurrently with another agent for treatment, although it is used alone for prophylaxis. Isoniazid is available for oral and parenteral administration. The commonly used total daily dose of isoniazid is 5 mg/kg, with a maximum of 300 mg; oral and intramuscular doses are identical. Isoniazid usually is given orally in a single daily dose but may be given in two divided doses. Although doses of 10 to 20 mg/kg, with a maximum of 600 mg, occasionally are used in severely ill patients, there is no evidence that this regimen is more effective. Children should receive 10 to 20 mg/kg per day (300 mg maximum). Isoniazid may be used as intermittent therapy for tuberculosis; after a minimum of 2 months of daily therapy of tuberculosis due to sensitive strains of M. tuberculosis with isoniazid, rifampin, and pyrazinamide, patients may be treated with twice-weekly doses of isoniazid (15 mg/kg orally) plus rifampin (10 mg/kg, up to 600 mg per dose) for 4 months. Pyridoxine (15 to 50 mg per day) should be administered with isoniazid to minimize adverse reactions (see below) in malnourished patients and those predisposed to neuropathy (e.g., the elderly, pregnant women, HIV-infected individuals, diabetics, alcoholics, and uremics; Snider, 1980). Untoward Effects The incidence of adverse reactions to isoniazid was estimated to be 5.4% among more than 2000 patients treated with the drug; the most prominent of these reactions were rash (2%), fever (1.2%), jaundice (0.6%), and peripheral neuritis (0.2%). Hypersensitivity to isoniazid may result in fever, various skin eruptions, hepatitis, and morbilliform, maculopapular, purpuric, and urticarial rashes. Hematological reactions also may occur (agranulocytosis, eosinophilia, thrombocytopenia, anemia). Vasculitis associated with antinuclear antibodies may appear during treatment but disappears when the drug is stopped. Arthritic symptoms (back pain; bilateral proximal interphalangeal joint involvement; arthralgia of the knees, elbows, and wrists; and the 'shoulder-hand' syndrome) have been attributed to this agent. If pyridoxine is not given concurrently, peripheral neuritis (most commonly paresthesia of feet and hands) is the most common reaction to isoniazid and occurs in about 2% of patients receiving 5 mg/kg of the drug daily. Higher doses may result in peripheral neuritis in 10% to 20% of patients. Neuropathy is more frequent in slow acetylators and in individuals with diabetes mellitus, poor nutrition, or anemia. The prophylactic administration of pyridoxine prevents the development not only of peripheral neuritis but also of most other nervous system disorders in practically all instances even when therapy lasts as long as 2 years. Isoniazid may precipitate convulsions in patients with seizure disorders and, rarely, in patients with no history of seizures. Optic neuritis and atrophy also have occurred during therapy with the drug. Muscle twitching, dizziness, ataxia, paresthesias, stupor, and toxic encephalopathy that may be fatal are other manifestations of the neurotoxicity of isoniazid. A number of mental abnormalities may appear during the use of this drug; among these are euphoria, transient impairment of memory, separation of ideas and reality, loss of self-control, and florid psychoses. Isoniazid is known to inhibit the parahydroxylation of phenytoin, and signs and symptoms of toxicity occur in approximately 27% of patients given both drugs, particularly in those who are slow acetylators (Miller et al., 1979). Concentrations of phenytoin in plasma should be monitored and adjusted if necessary. The dosage of isoniazid should not be changed. Although jaundice has been known for some time to be an untoward effect of exposure to isoniazid, not until the early 1970s did it become apparent that severe hepatic injury leading to death may occur in some individuals receiving this drug (Garibaldi et al., 1972). Additional studies in adults and children have confirmed this observation; the characteristic pathological process is bridging and multilobular necrosis. Continuation of the drug after symptoms of hepatic dysfunction have appeared tends to increase the severity of damage. The mechanisms responsible for this toxicity are unknown, although acetylhydrazine, which is a metabolite of isoniazid, causes hepatic damage in adults. Hence, patients who are rapid acetylators of isoniazid might be expected to be more likely to develop hepatotoxicity than slow acetylators. Whether or not this is true, however, is unresolved. A contributory role of alcoholic hepatitis has been noted, but chronic carriers of the hepatitis B virus tolerate isoniazid (McGlynn et al., 1986). Age appears to be the most important factor in determining the risk of isoniazid-induced hepatotoxicity. Hepatic damage is rare in patients less than 20 years old; the complication is observed in 0.3% of those 20 to 34 years old, and the incidence increases to 1.2% and 2.3% in individuals 35 to 49 and older than 50 years of age, respectively (Bass et al., 1994; Comstock, 1983). Up to 12% of patients receiving isoniazid may have elevated plasma aspartate and alanine transaminase activities (Bailey et al., 1974). Patients receiving isoniazid should be carefully evaluated at monthly intervals for symptoms of hepatitis (anorexia, malaise, fatigue, nausea, and jaundice) and warned to discontinue the drug if such symptoms occur. Some clinicians also prefer to determine serum aspartate aminotransferase activities at monthly intervals (Byrd et al., 1979) and recommend that an elevation greater than five times normal is cause for discontinuation of the drug. Most hepatitis occurs 4 to 8 weeks after the start of therapy. Isoniazid should be administered with great care to those with preexisting hepatic disease. Among miscellaneous reactions associated with isoniazid therapy are dryness of the mouth, epigastric distress, methemoglobinemia, tinnitus, and urinary retention. In persons predisposed to pyridoxine-deficiency anemia, the administration of isoniazid may result in its appearance in full-blown form. Treatment with large doses of the vitamin gradually returns the blood to normal in such cases (seeGoldman and Braman, 1972). A drug-induced syndrome resembling systemic lupus erythematosus has been reported. Overdose of isoniazid, as in attempted suicide, may result in nausea, vomiting, dizziness, slurred speech, and visual hallucinations followed by coma, seizures, metabolic acidosis, and hyperglycemia. Pyridoxine is an antidote in this setting; it should be given in a dose that approximates the amount of isoniazid ingested. Rifampin The rifamycins (rifampin, rifabutin, rifapentine) are a group of structurally similar, complex macrocyclic antibiotics produced by Streptomyces mediterranei (Farr, 2000); rifampin (RIFADIN RIMACTANE) is a semisynthetic derivative of one of theserifamycin B. Chemistry Rifampin is soluble in organic solvents and in water at acidic pH. It has the following structure:
Antibacterial Activity Rifampin inhibits the growth of most gram-positive bacteria as well as
many gram-negative microorganisms such as Escherichia coli, Pseudomonas,
indole-positive and indole-negative Proteus, and Klebsiella.
Rifampin is very active against Staphylococcus aureus and
coagulase-negative staphylococci; bactericidal concentrations range from 3 to
12 ng/ml. The drug also is highly active against Neisseria meningitidis
and Haemophilus influenzae; minimal inhibitory concentrations range
from 0.1 to 0.8 Rifampin in concentrations of 0.005 to 0.2 Bacterial Resistance Microorganisms, including mycobacteria, may develop resistance to rifampin rapidly in vitro as a one-step process, and one of every 107 to 108 tubercle bacilli is resistant to the drug. Resistance in most cases is due to mutations between codons 507 and 533 of the polymerase rpoB gene (Blanchard, 1996). This also appears to be the case in vivo, and therefore the antibiotic must not be used alone in the chemotherapy of tuberculosis. When rifampin has been used for eradication of the meningococcal carrier state, failures have been due to the appearance of drug-resistant bacteria after treatment for as little as 2 days. Microbial resistance to rifampin is due to an alteration of the target of this drug, DNA-dependent RNA polymerase. Certain rifampin-resistant bacterial mutants have decreased virulence. Tuberculosis caused by rifampin-resistant mycobacteria has been described in patients who had not received prior chemotherapy, but this is very rare (usually less than 1%; Cauthen et al., 1988). Mechanism of Action Rifampin inhibits DNA-dependent RNA polymerase of mycobacteria and
other microorganisms by forming a stable drugenzyme complex, leading to
suppression of initiation of chain formation (but not chain elongation) in
RNA synthesis. More specifically, the Absorption, Distribution, and Excretion The oral administration of rifampin produces peak concentrations in
plasma in 2 to 4 hours; after ingestion of 600 mg, this value is about 7 Following absorption from the gastrointestinal tract, rifampin is eliminated rapidly in the bile, and an enterohepatic circulation ensues. During this time, the drug is progressively deacetylated, to a degree that, after 6 hours, nearly all of the antibiotic in the bile is in the deacetylated form. This metabolite retains essentially full antibacterial activity. Intestinal reabsorption is reduced by deacetylation (as well as by food), and thus metabolism facilitates elimination of the drug. The half-life of rifampin varies from 1.5 to 5 hours and is increased in the presence of hepatic dysfunction; it may be decreased in patients receiving isoniazid concurrently who are slow inactivators of this drug. The half-life of rifampin is progressively shortened by about 40% during the first 14 days of treatment, owing to induction of hepatic microsomal enzymes with acceleration of deacetylation of the drug. Up to 30% of a dose of the drug is excreted in the urine and 60% to 65% in the feces; less than half of this may be unaltered antibiotic. Adjustment of dosage is not necessary in patients with impaired renal function. Rifampin is distributed throughout the body and is present in effective concentrations in many organs and body fluids, including the CSF (Sippel et al., 1974). This is perhaps best exemplified by the fact that the drug may impart an orange-red color to the urine, feces, saliva, sputum, tears, and sweat; patients should be so warned. (For various aspects of rifampin metabolism, seeFuresz, 1970; Farr, 2000.) Therapeutic Uses Rifampin is available alone and as a fixed-dose combination with isoniazid (150 mg of isoniazid, 300 mg of rifampin; RIFAMATE) or with isoniazid and pyrazinamide (50 mg of isoniazid, 120 mg of rifampin, and 300 mg pyrazinamide; RIFATER). Rifampin and isoniazid are the most effective drugs available for the treatment of tuberculosis. The dose of rifampin for treatment of tuberculosis in adults is 600 mg, given once daily, either 1 hour before or 2 hours after a meal. Children should receive 10 mg/kg, with a daily maximum, given in the same way. Doses of 15 mg/kg or higher are associated with increased hepatotoxicity in children (Centers for Disease Control, 1980). Rifampin, like isoniazid, should never be used alone for this disease because of the rapidity with which resistance may develop. Despite the long list of untoward effects from rifampin, their incidence is low, and treatment seldom has to be interrupted. The use of rifampin in the chemotherapy of tuberculosis is detailed below. Rifampin also is indicated for the prophylaxis of meningococcal disease and H. influenzae meningitis. To prevent meningococcal disease, adults may be treated with 600 mg twice daily for 2 days or 600 mg once daily for 4 days; children should receive 10 to 20 mg/kg, to a maximum of 600 mg. Untoward Effects Rifampin generally is well tolerated. When given in usual doses, fewer than 4% of patients with tuberculosis have significant adverse reactions; the most common are rash (0.8%), fever (0.5%), and nausea and vomiting (1.5%) (seeGrosset and Leventis, 1983). Rarely, hepatitis and deaths due to liver failure have been observed in patients who received other hepatoxic agents in addition to rifampin or who had preexisting liver disease. Hepatitis from rifampin rarely occurs in patients with normal hepatic function; likewise, the combination of isoniazid and rifampin appears generally safe in such patients (Gangadharam, 1986). However, chronic liver disease, alcoholism, and old age appear to increase the incidence of severe hepatic problems when rifampin is given alone or concurrently with isoniazid. Administration of rifampin on an intermittent schedule (less than twice weekly) and/or daily doses of 1200 mg or greater is associated with frequent side effects, and the drug should not be used in this manner. A flulike syndrome with fever, chills, and myalgias develops in 20% of patients so treated. The syndrome also may include eosinophilia, interstitial nephritis, acute tubular necrosis, thrombocytopenia, hemolytic anemia, and shock (Girling and Hitze, 1979). Because rifampin is a potent inducer of hepatic microsomal enzymes, its administration results in a decreased half-life for a number of compounds, including HIV protease and nonnucleoside reverse transcriptase inhibitors, digitoxin, digoxin, quinidine, disopyramide, mexiletine, tocainide, ketoconazole, propranolol, metoprolol, clofibrate, verapamil, methadone, cyclosporine, corticosteroids, oral anticoagulants, theophylline, barbiturates, oral contraceptives, halothane, fluconazole, and the sulfonylureas (Farr, 2000). Rifabutin has less of an effect on the metabolism of the HIV protease inhibitors indinavir and nelfinavir. The significant interaction between rifampin and oral anticoagulants of the coumarin type leads to a decrease in efficacy of these agents. This effect appears about 5 to 8 days after rifampin administration is started and persists for 5 to 7 days after it is stopped (O'Reilly, 1975). The ability of rifampin to enhance the catabolism of a variety of steroids leads to the decreased effectiveness of oral contraceptives (Skolnick et al., 1976). The increased metabolism of methadone has led to reports of precipitation of withdrawal syndromes. Rifampin may reduce biliary excretion of contrast media used for visualization of the gallbladder (seeBaciewicz et al., 1987). Gastrointestinal disturbances produced by rifampin (epigastric distress, nausea, vomiting, abdominal cramps, diarrhea) have occasionally required discontinuation of the drug. Various symptoms related to the nervous system also have been noted, including fatigue, drowsiness, headache, dizziness, ataxia, confusion, inability to concentrate, generalized numbness, pain in the extremities, and muscular weakness. Among hypersensitivity reactions are fever, pruritus, urticaria, various types of skin eruptions, eosinophilia, and soreness of the mouth and tongue. Hemolysis, hemoglobinuria, hematuria, renal insufficiency, and acute renal failure have been observed rarely; these also are thought to be hypersensitivity reactions. Thrombocytopenia, transient leukopenia, and anemia have occurred during therapy. Since the potential teratogenicity of rifampin is unknown and the drug is known to cross the placenta, it is best to avoid the use of this agent during pregnancy. Graber and associates (1973) have noted immunoglobulin light-chain proteinuria (either kappa, lambda, or both) in about 85% of patients with tuberculosis treated with rifampin. None of the patients had symptoms or electrophoretic patterns compatible with myeloma. However, renal failure has been associated with light-chain proteinuria (Warrington et al., 1977). Rifampin is a drug of choice for chemoprophylaxis of meningococcal
disease and meningitis due to H. influenzae in household contacts of
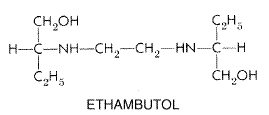
patients with such infections. Combined with a Ethambutol Chemistry Ethambutol is a water-soluble and heat-stable compound. The structural formula is as follows:
Antibacterial Activity Nearly all strains of M. tuberculosis and M. kansasii as well as a number of strains of M. avium complex are sensitive to ethambutol (Pablos-Mndez et al., 1998). The sensitivities of other nontuberculous organisms are variable. Ethambutol has no effect on other bacteria. It suppresses the growth of most isoniazid- and streptomycin-resistant tubercle bacilli. Resistance to ethambutol develops very slowly in vitro. Mycobacteria take up ethambutol rapidly when the drug is added to cultures that are in the exponential growth phase. However, growth is not significantly inhibited before about 24 hours; the drug is tuberculostatic. Ethambutol blocks arabinosyl transferases involved in cell wall biosynthesis (Takayama et al., 1979). Bacterial resistance to the drug develops in vivo via single amino acid changes in the embA gene when ethambutol is given in the absence of another effective agent (Belanger et al., 1996). Absorption, Distribution, and Excretion About 75% to 80% of an orally administered dose of ethambutol is
absorbed from the gastrointestinal tract. Concentrations in plasma are
maximal in human beings 2 to 4 hours after the drug is taken and are
proportional to the dose. A single dose of 25 mg/kg produces a plasma
concentration of 2 to 5 Within 24 hours, three-fourths of an ingested dose of ethambutol is excreted unchanged in the urine; up to 15% is excreted in the form of two metabolites, an aldehyde and a dicarboxylic acid derivative. Renal clearance of ethambutol is approximately 7 mlmin1 kg1; thus it is evident that the drug is excreted by tubular secretion in addition to glomerular filtration. Therapeutic Uses Ethambutol (ethambutol hydrochloride; MYAMBUTOL) has been used with notable success in the therapy of tuberculosis of various forms when given concurrently with isoniazid. Because of a lower incidence of toxic effects and better acceptance by patients, ethambutol has essentially replaced aminosalicylic acid (seeBobrowitz, 1974). Ethambutol is available for oral administration in tablets containing the d isomer. The usual adult dose of ethambutol is 15 mg/kg given once a day. Some physicians prefer to institute therapy with a dose of 25 mg/kg per day for the first 60 days and then to reduce the dose to 15 mg/kg per day, particularly for those who have received previous therapy. Ethambutol accumulates in patients with impaired renal function, and adjustment of dosage is necessary. Ethambutol is not recommended for children under 5 years of age, in part because of concern about the ability to test their visual acuity (see below). Children from ages 6 to 12 years should receive 10 to 15 mg/kg per day. The use of ethambutol in the chemotherapy of tuberculosis is described below. Untoward Effects The most important side effect is optic neuritis, resulting in decrease of visual acuity and loss of ability to differentiate red from green. The incidence of this reaction is proportional to the dose of ethambutol and is observed in 15% of patients receiving 50 mg/kg per day, in 5% of patients receiving 25 mg/kg per day, and in fewer than 1% of patients receiving daily doses of 15 mg/kg (the recommended dose for treatment of tuberculosis). The intensity of the visual difficulty is related to the duration of therapy after the decrease in visual acuity first becomes apparent, and it may be unilateral or bilateral. Tests of visual acuity and red-green discrimination prior to the start of therapy and periodically thereafter are thus recommended. Recovery usually occurs when ethambutol is withdrawn; the time required is a function of the degree of visual impairment. Ethambutol produces very few reactions. Daily doses of 15 mg/kg are minimally toxic. Fewer than 2% of nearly 2000 patients who received 15 mg/kg of ethambutol had adverse reactions; 0.8% experienced diminished visual acuity, 0.5% had a rash, and 0.3% developed drug fever. Other side effects that have been observed are pruritus, joint pain, gastrointestinal upset, abdominal pain, malaise, headache, dizziness, mental confusion, disorientation, and possible hallucination. Numbness and tingling of the fingers owing to peripheral neuritis are infrequent. Anaphylaxis and leukopenia are rare. Therapy with ethambutol results in an increased concentration of urate in the blood in about 50% of patients, owing to decreased renal excretion of uric acid. The effect may be detectable as early as 24 hours after a single dose or as late as 90 days after treatment is started. This untoward effect is possibly enhanced by isoniazid and pyridoxine (Postlethwaite et al., 1972). Streptomycin A discussion of the pharmacology of streptomycin, including its adverse effects and its uses in infections other than tuberculosis, is presented in Chapter 46: Antimicrobial Agents: The Aminoglycosides. Only features of the drug related to its antibacterial activity and therapeutic effects in the management of diseases caused by mycobacteria are considered here. History Streptomycin was the first clinically effective drug to become available for the treatment of tuberculosis. At first, it was given in large doses, but problems related to toxicity and the development of resistant microorganisms seriously limited its usefulness. The antibiotic was then administered in smaller quantities, but streptomycin administered alone still proved to be far from the ideal agent for the management of all forms of the disease. However, the discovery of other compounds that, given concurrently with the antibiotic, reduced the rate at which microorganisms became drug-resistant enabled physicians to treat tuberculosis effectively with streptomycin. It is now the least used of the 'first-line' agents in the therapy of tuberculosis. Antibacterial Activity Streptomycin is bactericidal for the tubercle bacillus in vitro.
Concentrations as low as 0.4 The activity of streptomycin in vivo is essentially suppressive. When the antibiotic is administered to experimental animals prior to inoculation with the tubercle bacillus, the development of disease is not prevented. Infection progresses until the animals' immunological mechanisms respond. The presence of viable microorganisms in abscesses and in the regional lymph nodes adds support to the concept that the activity of streptomycin in vivo is to suppress, not to eradicate, the tubercle bacillus. This property of streptomycin may be related to the observation that the drug does not readily enter living cells and thus cannot kill intracellular microbes. Bacterial Resistance Large populations of all strains of tubercle bacilli include a number of cells that are markedly resistant to streptomycin because of mutation. However, primary resistance to the antibiotic is found in only 2% to 3% of isolates of M. tuberculosis. Selection for resistant tubercle bacilli occurs in vivo as it
does in vitro. In general, the longer therapy is continued, the
greater is the incidence of resistance to streptomycin. When streptomycin was
used alone, as many as 80% of patients harbored insensitive tubercle bacilli
after 4 months of treatment; many of these microorganisms were not inhibited
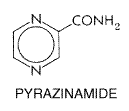
by concentrations of drug as high as 1000 Therapeutic Uses Since other effective agents have become available, the use of streptomycin for the treatment of pulmonary tuberculosis has been sharply reduced. Many clinicians prefer to give four drugs, of which streptomycin may be one, for the most serious forms of tuberculosis, such as disseminated disease or meningitis. For tuberculosis, adults should be given 15 mg/kg per day in divided doses given every 12 hours, not to exceed 1 g per day. Children should receive 20 to 40 mg/kg per day in divided doses every 12 to 24 hours, not to exceed 1 g per day. Therapy usually is discontinued after 2 to 3 months, or sooner if cultures become negative. Dosage schedules for various forms of tuberculosis are discussed further below. Untoward Effects Untoward effects of streptomycin are considered in detail in Chapter 46: Antimicrobial Agents: The Aminoglycosides. Of 515 patients with tuberculosis who were treated with this aminoglycoside, 8.2% had adverse reactions; half of these involved the auditory and vestibular functions of the eighth cranial nerve. Other relatively frequent problems included rash (in 2%) and fever (in 1.4%). Pyrazinamide Chemistry Pyrazinamide is the synthetic pyrazine analog of nicotinamide. It has the following structural formula:
Antibacterial Activity Pyrazinamide exhibits bactericidal activity in vitro only at a
slightly acidic pH. Activity at acid pH is ideal, since M. tuberculosis
resides in an acidic phagosome within the macrophage (Jacobs, 2000). Tubercle
bacilli within monocytes in vitro are inhibited or killed by the drug
at a concentration of 12.5 Absorption, Distribution, and Excretion Pyrazinamide is well absorbed from the gastrointestinal tract, and it
is widely distributed throughout the body. The oral administration of 500 mg
produces plasma concentrations of about 9 to 12 Therapeutic Uses Pyrazinamide has become an important component of short-term (6-month) multiple-drug therapy of tuberculosis (British Thoracic Association, 1983; Bass et al., 1994). Pyrazinamide is available in tablets for oral administration. The daily dose for adults is 15 to 30 mg/kg orally, given as a single dose. The maximum quantity to be given is 2 g per day, regardless of weight. Children should receive 15 to 30 mg/kg per day; daily doses should not exceed 2 g. Pyrazinamide also has been safe and effective when administered twice or thrice weekly (at increased dosages). Untoward Effects Injury to the liver is the most serious side effect of pyrazinamide. When a dose of 40 to 50 mg/kg is administered orally, signs and symptoms of hepatic disease appear in about 15% of patients, with jaundice in 2% to 3% and death due to hepatic necrosis in rare instances. Elevations of the plasma alanine and aspartate aminotransferases are the earliest abnormalities produced by the drug. Regimens employed currently (15 to 30 mg/kg per day) are much safer (Girling, 1978). All patients who are being treated with pyrazinamide should undergo studies of hepatic function before the drug is administered; these studies should be repeated at frequent intervals during the entire period of treatment. If evidence of significant hepatic damage becomes apparent, therapy must be stopped. Pyrazinamide should not be given to individuals with any degree of hepatic dysfunction unless this is absolutely unavoidable. The drug inhibits excretion of urate, resulting in hyperuricemia in nearly
all patients; acute episodes of gout have occurred. Other untoward effects
that have been observed with pyrazinamide are arthralgias, anorexia, nausea
and vomiting, dysuria, malaise, and fever. While some international
organizations recommend the use of pyrazinamide in pregnancy, this is not the
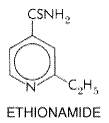
case in the Ethionamide Chemistry Synthesis and study of a variety of congeners of thioisonicotinamide revealed that an alpha-ethyl derivativeethionamide (TRECATOR-SC)is considerably more effective than the parent compound. It has the following structural formula:
Antibacterial Activity The multiplication of M. tuberculosis is suppressed by
concentrations of ethionamide ranging from 0.6 to 2.5 Absorption, Distribution, and Excretion The oral administration of 1 g of ethionamide yields peak
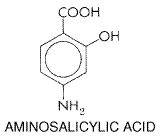
concentrations in plasma of about 20 Therapeutic Uses Ethionamide is a secondary agent, to be used concurrently with other drugs only when therapy with primary agents is ineffective or contraindicated. Ethionamide is administered only orally. The initial dosage of ethionamide for adults is 250 mg twice daily; it is increased by 125 mg per day every 5 days until a dose of 15 to 20 mg/kg per day is achieved. The maximal dose is 1 g daily. The drug is best taken with meals in divided doses in order to minimize gastric irritation. Children should receive 15 to 20 mg/kg per day in two divided doses, not to exceed 1 g per day. Untoward Effects The most common reactions to ethionamide are anorexia, nausea, and vomiting. A metallic taste also may be noted. Severe postural hypotension, mental depression, drowsiness, and asthenia are common. Convulsions and peripheral neuropathy are rare. Other reactions referable to the nervous system include olfactory disturbances, blurred vision, diplopia, dizziness, paresthesias, headache, restlessness, and tremors. Severe allergic skin rashes, purpura, stomatitis, gynecomastia, impotence, menorrhagia, acne, and alopecia also have been observed. Hepatitis has been associated with the use of the drug in about 5% of cases (Simon et al., 1969). The signs and symptoms of hepatotoxicity clear when treatment is stopped. Hepatic function should be assessed at regular intervals in patients receiving ethionamide. The concomitant use of pyridoxine is recommended for patients being treated with ethionamide. Aminosalicylic Acid Chemistry The structural formula of aminosalicylic acid (p-aminosalicylic acid, PAS) is as follows:
Antibacterial Activity Aminosalicylic acid is bacteriostatic. In vitro, most strains
of M. tuberculosis are sensitive to a concentration of 1 Studies of the treatment of experimental M. tuberculosis infections indicate that aminosalicylic acid exerts a beneficial effect on the disease. However, the doses required are relatively large, and the compound must be present continuously. Aminosalicylic acid alone is of little value in the treatment of tuberculosis in human beings. Bacterial Resistance Strains of tubercle bacilli insensitive to several hundred times the usual bacteriostatic concentration of aminosalicylic acid can be produced in vitro. Resistant strains of tubercle bacilli also emerge in patients treated with aminosalicylic acid, but much more slowly than with streptomycin. Mechanism of Action Aminosalicylic acid is a structural analog of paraaminobenzoic acid, and its mechanism of action appears to be very similar to that of the sulfonamides (seeChapter 44: Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections). Since the sulfonamides are ineffective against M. tuberculosis, and aminosalicylic acid is inactive against sulfonamide-susceptible bacteria, it is probable that the enzymes responsible for folate biosynthesis in various microorganisms may be quite exacting in their capacity to distinguish various analogs from the true metabolite. Absorption, Distribution, and Excretion Aminosalicylic acid is readily absorbed from the gastrointestinal
tract. A single oral dose of 4 g of the free acid produces maximal
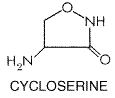
concentrations in plasma of about 75 The drug has a half-life of about 1 hour, and concentrations in plasma are negligible within 4 to 5 hours after a single conventional dose. Over 80% of the drug is excreted in the urine; more than 50% is in the form of the acetylated compound. The largest portion of the remainder is made up of the free acid. Excretion of aminosalicylic acid is greatly retarded in the presence of renal dysfunction, and the use of the drug is not recommended in such patients. Probenecid decreases the renal excretion of this agent. Therapeutic Uses Aminosalicylic acid is a 'second-line' agent. Its importance in the management of pulmonary and other forms of tuberculosis has markedly decreased since more active and better-tolerated drugs, such as rifampin and ethambutol, have been developed (seeChemotherapy of Tuberculosis, below). Aminosalicylic acid is administered orally in a daily dose of 10 to 12 g. Because it is a gastric irritant, the drug is best administered after meals, the daily dose being divided into two to four equal portions. Children should receive 150 to 300 mg/kg per day in three to four divided doses. Untoward Effects The incidence of untoward effects associated with the use of aminosalicylic acid is approximately 10% to 30%. Gastrointestinal problemsincluding anorexia, nausea, epigastric pain, abdominal distress, and diarrheaare predominant, and patients with peptic ulcer tolerate the drug poorly. Compliance is often poor because of gastrointestinal distress. Hypersensitivity reactions to aminosalicylic acid are seen in 5% to 10% of patients. High fever may develop abruptly, with intermittent spiking, or it may appear gradually and be low-grade. Generalized malaise, joint pains, or sore throat may be present at the same time. Skin eruptions of various types appear as isolated reactions or accompany the fever. Among the hematological abnormalities that have been observed are leukopenia, agranulocytosis, eosinophilia, lymphocytosis, an atypical mononucleosis syndrome, and thrombocytopenia. Acute hemolytic anemia may appear in some instances. Cycloserine Cycloserine SEROMYCIN) is a broad-spectrum antibiotic produced by Streptococcus orchidaceus. It was first isolated from a fermentation brew in 1955 and was later synthesized. Currently, cycloserine is used in conjunction with other tuberculostatic drugs in the treatment of pulmonary or extrapulmonary tuberculosis when primary agents (isoniazid, rifampin, ethambutol, pyrazinamide, streptomycin) have failed. Chemistry Cycloserine is D-4-amino-3-isoxazolidone; the structural formula is as follows:
The drug is stable in alkaline solution but is rapidly destroyed when exposed to neutral or acidic pH. Antibacterial Activity and Mechanism of Action Cycloserine is inhibitory for M. tuberculosis in concentrations
of 5 to 20 Absorption, Distribution, and Excretion When given orally, 70% to 90% of cycloserine is rapidly absorbed. Peak
concentrations in plasma are reached 3 to 4 hours after a single dose and are
in the range of 20 to 35 Therapeutic Uses Cycloserine should be used only when retreatment is necessary or when microorganisms are resistant to other drugs. When cycloserine is employed to treat tuberculosis, it must be given together with other effective agents. Cycloserine is available for oral administration. The usual dose for adults is 250 to 500 mg twice daily. Untoward Effects Reactions to cycloserine most commonly involve the central nervous system. They tend to appear within the first 2 weeks of therapy and usually disappear when the drug is withdrawn. Among the central manifestations are somnolence, headache, tremor, dysarthria, vertigo, confusion, nervousness, irritability, psychotic states with suicidal tendencies, paranoid reactions, catatonic and depressed reactions, twitching, ankle clonus, hyperreflexia, visual disturbances, paresis, and tonic-clonic or absence seizures. Large doses of cycloserine or the ingestion of ethyl alcohol increases the risk of seizures. Cycloserine is contraindicated in individuals with a history of epilepsy. Other Drugs The agents grouped in this section are similar in several aspects. They are all 'second-line' drugs that are used only for treatment of disease caused by resistant microorganisms or by nontuberculous mycobacteria. They all must be given parenterally, and they have similar pharmacokinetics and toxicity. Since these agents are potentially ototoxic and nephrotoxic, no two drugs from this group should be employed simultaneously, and these drugs should not be used in combination with streptomycin. Kanamycin,
an aminoglycoside that is discussed in Chapter 46: Antimicrobial Agents: The Aminoglycosides, inhibits the growth of M.
tuberculosisin vitro in a concentration of 10 Amikacin also is an aminoglycoside (seeChapter 46: Antimicrobial Agents: The Aminoglycosides). It is extremely active against several mycobacterial species and has a role in the treatment of disease caused by nontuberculous mycobacteria (seeBrown and Wallace, 2000). Capreomycin CAPASTAT SULFATE) is an antimycobacterial cyclic peptide elaborated by Streptococcus capreolus. It consists of four active componentscapreomycins IA, IB, IIA, and IIB. The agent used clinically contains primarily IA and IB. Bacterial resistance to capreomycin develops when it is given alone; such microorganisms show cross-resistance with kanamycin and neomycin. Capreomycin is used only in conjunction with other appropriate antitubercular drugs in treatment of pulmonary tuberculosis when bactericidal agents cannot be tolerated or when causative organisms have become resistant. Capreomycin must be given intramuscularly. The recommended daily dose is 15 to 30 mg/kg per day or up to 1 g for 60 to 120 days, followed by 1 g two to three times a week. The reactions associated with the use of capreomycin are hearing loss, tinnitus, transient proteinuria, cylindruria, and nitrogen retention. Severe renal failure is rare. Eosinophilia is common. Leukocytosis, leukopenia, rashes, and fever have also been observed. Injections of the drug may be painful. Chemotherapy of Tuberculosis The availability of effective agents has so altered the treatment of tuberculosis that most patients are now treated in the ambulatory setting, often after diagnosis and initial therapy in a general hospital. Prolonged bed rest is not necessary or even helpful in speeding recovery. Patients must be seen at frequent intervals to follow the course of their disease and treatment. The local health department must be notified of all cases. Contacts should be investigated for the possibility of disease and for the appropriateness of prophylactic therapy with isoniazid. The majority of cases of previously untreated tuberculosis in the Drug interactions are a special concern in patients receiving highly active antiretroviral therapy. The rifamycins accelerate the metabolism of protease inhibitors and nonnucleoside reverse transcriptase inhibitors. Of the rifamycins, rifabutin has the least effect on indinavir and nelfinavir serum levels. Patients infected with the human immunodeficiency virus (HIV) may
benefit from longer (9- to 12-month) treatment regimens (Havlir and Barnes,
1999). Treatment should be initiated with at least a four-drug regimen
consisting of isoniazid, rifabutin, pyrazinamide, and ethambutol or streptomycin.
In patients with a high likelihood of infection with multidrug-resistant
strains, an initial five- or six-drug regimen may be appropriate (Lane et
al., 1994; Gallant et al., 1994). Treatment should be continued
for at least 6 months after three negative cultures have been obtained. If
isoniazid or rifampin cannot be used, therapy should be continued for at
least 18 months (12 months after cultures become negative). Chemoprophylaxis
(seeChemoprophylaxis of Tuberculosis) should be undertaken if a
patient with HIV infection has a positive tuberculin test (induration Therapy of Specific Types of Tuberculosis Therapy for uncomplicated drug-sensitive pulmonary tuberculosis consists of isoniazid (5 mg/kg, up to 300 mg per day), rifampin (10 mg/kg per day, up to 600 mg daily), and pyrazinamide (15 to 30 mg/kg per day or a maximum of 2 g per day). Pyridoxine, 15 to 50 mg per day, also should be included for most adults to minimize adverse reactions to isoniazid (Snider, 1980). Isoniazid, rifampin, and pyrazinamide are given for 2 months; isoniazid and rifampin are then continued for 4 additional months. Children are treated similarly; doses are isoniazid, 10 mg/kg per day (300 mg maximum); rifampin, 10 to 20 mg/kg per day (600 mg maximum); pyrazinamide, 15 to 30 mg/kg per day (2 g maximum; Bass et al., 1994). Surgery is rarely indicated (Haas, 2000). The multidrug regimen of isoniazid, rifampin, and ethambutol is considered safe during pregnancy. Certain patients should receive at least four drugs initially to ensure that the microorganisms will be susceptible to at least two of the agents. These patients include (1) those known to have been exposed to drug-resistant microorganisms; (2) Asians and Hispanics, especially if they are recent immigrants; (3) those with miliary tuberculosis or other extrapulmonary disease; (4) those with meningitis; (5) those with extensive pulmonary disease; and (6) those with HIV infection. The microorganisms should be cultured for determination of their sensitivity to antimicrobial agents, but results will not be available for several weeks. The fourth agent may be either ethambutol (usual adult dose of 15 mg/kg once per day) or streptomycin (1 g daily). The dosage of streptomycin is reduced to 1 g twice weekly after 2 months. Some physicians prefer to institute ethambutol therapy with a dose of 25 mg/kg per day for the first 60 days and then to reduce the dose to 15 mg/kg per day, particularly for those who have received previous therapy. Clinical improvement is readily discernible in the vast majority of patients with pulmonary tuberculosis if the treatment is appropriate. Efficacy usually becomes obvious within the first 2 weeks of therapy and is evidenced by a reduction of fever, decrease in cough, gain in weight, and increase in the sense of well-being. Progressive radiological improvement also is evident. Over 90% of patients who receive optimal treatment will have negative cultures within 3 to 6 months, depending on the severity of the disease. Cultures that remain positive after 6 months frequently yield resistant microorganisms; the value of using an alternative therapeutic program should then be considered. Failure of chemotherapy may be due to (1) irregular or inadequate therapy (resulting in persistent or resistant mycobacteria) caused by poor patient compliance during the protracted therapeutic regimen; (2) the use of a single drug, with interruption necessitated by toxicity or hypersensitivity; (3) an inadequate initial regimen; or (4) the primary resistance of the microorganism. Problems in Chemotherapy Bacterial Resistance to Drugs One of the more important problems in the chemotherapy of tuberculosis is bacterial resistance. The primary reason for development of drug resistance is poor patient compliance. To prevent noncompliance and the attendant development of drug-resistant tuberculosis, directly observed therapy is advisable for most patients. For directly observed therapy, a health care provider observes the patient ingest the medications 2 to 5 times weekly (Barnes and Barrows, 1993; Chaulk and Kazandjian, 1998). Where drug resistance is suspected but sensitivities are not yet known (as in patients who have undergone several courses of treatment), therapy should be instituted with five or six drugs, including two or three that the patient has not received in the past. Such a program might include isoniazid, rifampin, ethambutol, streptomycin, pyrazinamide, and ethionamide. Some physicians include isoniazid in the therapeutic regimen, even if microorganisms are resistant, because of some evidence that disease with isoniazid-resistant mycobacteria does not 'progress' during such therapy. Others prefer to discontinue isoniazid to lessen the possibility of toxicity. Therapy should be continued for at least 24 months. Nontuberculous (Atypical) Mycobacteria These microorganisms (not including M. avium complex, which is discussed later) have been recovered from a variety of lesions in human beings (Brown and Wallace, 2000). Because they frequently are resistant to many of the commonly used agents, they must be examined for sensitivity in vitro and drug therapy selected on this basis. In some instances, surgical removal of the infected tissue followed by long-term treatment with effective agents is necessary. M. kansasii causes disease similar to that caused by M. tuberculosis, but it may be milder. The microorganisms may be resistant to isoniazid. Therapy with isoniazid, rifampin, and ethambutol has been successful (Pezzia et al., 1981; Lane et al., 1994). M. marinum causes skin lesions. A combination of rifampin and ethambutol is probably effective; minocycline (Loria, 1976) or tetracycline is active in vitro and is used by some physicians (Izumi et al., 1977). M. scrofulaceum is an uncommon cause of cervical lymphadenitis. Surgical excision still seems to be the therapy of choice (Lincoln and Gilberg, 1972). Microbes of the M. fortuitum complex (including Mycobacterium chelonei) are usually saprophytes, but they may cause chronic lung disease and infections of skin and soft tissues. The microorganisms are highly resistant to most drugs, but amikacin, cefoxitin, and tetracyclines are active in vitro (Sanders et al., 1977; Sanders, 1982). Chemoprophylaxis of Tuberculosis There are several approaches to the chemoprophylaxis of tuberculosis. The classical prophylaxis with 12 months of isoniazid resulted in a 75% reduction in the risk of active tuberculosis (from an incidence of 14.3% to 3.6% over 5 years). A 6-month course of isoniazid therapy was nearly as effective, with a 65% risk reduction and a lower incidence of isoniazid-induced hepatitis (IUAT, 1982). Recently, a 2-month regimen of daily rifampin and pyrazinamide was shown to be as effective as 12 months of isoniazid in one study of HIV-infected individuals (Halsey et al., 1998). Prophylactic therapy can effectively prevent the development of active tuberculosis in certain instances (Haas, 2000). There are three categories of patients for whom prophylactic therapy should be considered: those exposed to tuberculosis but who have no evidence of infection; those with infection [positive tuberculin test: more than 5 mm (HIV infected) or 10 mm (not immunocompromised) of induration to 5 units of purified protein derivative (PPD)] and no apparent disease; and those with a history of tuberculosis but in whom the disease is presently 'inactive' (seeBass et al., 1994; Stead and To, 1987; Wilkinson et al., 1998; Gordin et al., 1997; Gallant et al., 1994). The main risk of chemoprophylaxis is isoniazid-induced hepatitis, which is much more common about the age of 35. Some authorities argue that monitored isoniazid prophylaxis minimizes the risk of toxicity even in patients over the age of 35 (Salpeter et al., 1997). Household contacts and other close associates of patients with tuberculosis who have negative tuberculin tests should receive isoniazid for at least 6 months after the contact has been broken, regardless of age. This is especially important for children. If the tuberculin skin test becomes positive, therapy should be continued for 12 months. Persons without apparent disease whose skin test has converted from negative to positive within the preceding 2 years should probably receive isoniazid for 12 months regardless of age. These patients are considered to be 'infected' but not to have clinical disease. Some authorities believe that persons with positive skin tests, no matter when they became so, who are under 35 years of age, or who are at risk of infection because of such factors as infection with HIV, immunosuppressive therapy, leukemia, lymphoma, or silicosis should receive isoniazid for 1 year (Bass et al., 1994; Gallant et al., 1994). For individuals over 35 years of age, the risk of isoniazid toxicity may outweigh the potential benefit of therapy. Patients with old 'inactive' tuberculosis who have not received adequate chemotherapy in the past should be considered for 1 year of treatment with isoniazid (seeComstock, 1983). HIV-infected intravenous drug abusers with a positive skin test have an approximately 8% chance per year of developing active tuberculosis (Selwyn et al., 1989). Isoniazid prophylaxis in HIV-infected persons appears to be as effective as in nonimmunocompromised persons (Wilkinson et al., 1998). The CDC recommends that isoniazid prophylaxis be continued for 12 months. Prophylaxis should be given to HIV-infected persons with more than 5 mm of induration to 5 units of PPD. A 1991 CDC recommendation that anergic HIV-infected persons from populations at risk for tuberculosis receive prophylaxis was reversed in 1997, when it was shown to be without foundation (Gordin et al., 1997; Whalen et al., 1997). Persons infected with HIV who are exposed to multidrug-resistant tuberculosis should receive prophylaxis with high-dose ethambutol and pyrazinamide, with or without a fluoroquinolone (Gallant et al., 1994). Prophylaxis with isoniazid is contraindicated for patients who have active hepatic disease or who have had reactions to the drug. The recent demonstration of the comparable efficacy of a 2-month course of rifampin and pyrazinamide (in HIV-infected adults) offers a potential alternative to isoniazid prophylaxis (Halsey et al., 1998). In pregnant women, prophylaxis usually should be delayed until after delivery. For prophylaxis, isoniazid generally is given to adults in a daily dose of 300 mg. Children should receive 10 mg/kg to a maximal daily dose of 300 mg. |
Drugs for Mycobacterium avium Complex
|
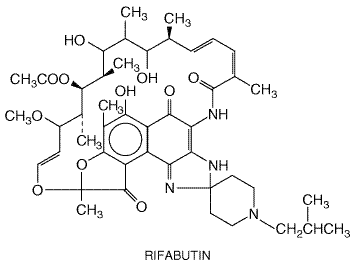
Before the advent of highly active antiretroviral therapy (HAART, seeChapter 51: Antiretroviral Agents: Antiretroviral Agents) and the use of prophylactic regimens, disseminated infection with M. avium complex (MAC) bacteria occurred in 15% to 40% of patients with HIV infection. Infections with MAC now are greatly reduced (Benson, 199798). Patients with M. avium complex infection usually are in advanced stages of HIV disease, with CD4 T-lymphocyte counts below 100/mm3 and symptoms of fever, night sweats, weight loss, and anemia at the time of diagnosis (Masur et al., 1993). In non-HIV-infected persons, MAC infection usually is limited to the lungs and presents with a chronic productive cough and chest roentgenograms showing evidence of limited, diffuse, and/or cavitary disease (Havlir and Ellner, 2000). Although standard antimycobacterial agents have little activity against MAC, new antimicrobial agents with activity against MAC recently have become available. These agents currently are in use for both the prevention and treatment of MAC in patients with AIDS. Rifabutin Rifabutin MYCOBUTIN) is a derivative of rifamycin S. Rifabutin shares its mechanism of action with rifampin (inhibition of mycobacterial RNA polymerase), but is more active in vitro and in experimental murine tuberculosis than is rifampin. Chemistry Rifabutin is soluble in organic solvents and at low concentrations (0.19 mg/ml) in water. It has the following structure:
Antibacterial Activity Rifabutin has better activity against the MAC organisms than does rifampin.
Rifabutin is active in vitro against MAC bacteria isolated from both
HIV-infected (where the majority of MAC infections are M. avium) and
non-HIV-infected individuals (where approximately 40% of MAC infections are M.
intracellulare). Rifabutin inhibits the growth of most MAC isolates at
concentrations ranging from 0.25 to 1.0 Bacterial Resistance Cross-resistance between rifampin and rifabutin occurs to some extent
with both M. avium and M. tuberculosis; of 225 M. avium
strains that were resistant to 10 Absorption, Distribution, and Excretion The oral administration of 300 mg of rifabutin produces a peak plasma
concentration of approximately 0.4 Therapeutic Uses Rifabutin is effective for the prevention of MAC infection in HIV-infected individuals. At a dose of 300 mg per day, rifabutin decreased the frequency of MAC bacteremia by half (Nightingale et al., 1993). However, azithromycin or clarithromycin are more effective and less likely to interact with HAART drugs. Rifabutin also is commonly substituted for rifampin in the treatment of tuberculosis in HIV-infected patients, as it has a less profound interaction with indinavir and nelfinavir (Haas, 2000). Rifabutin also is used in combination with clarithromycin and ethambutol for the therapy of MAC disease (Shafran et al., 1996). Untoward Effects Rifabutin generally is well tolerated in persons with HIV infection; primary reasons for discontinuation of therapy include rash (4%), gastrointestinal intolerance (3%), and neutropenia (2%; Nightingale et al., 1993). Overall neutropenia occurred in 25% of patients with severe HIV infection who received rifabutin. Uveitis and arthralgias have occurred in patients receiving rifabutin doses greater than 450 mg daily in combination with clarithromycin or fluconazole. Patients should be cautioned to discontinue the drug if visual symptoms (pain or blurred vision) occur. Like rifampin, the drug causes an orange-tan discoloration of skin, urine, feces, saliva, tears, and contact lenses. Rarely, thrombocytopenia, a flulike syndrome, hemolysis, myositis, chest pain, and hepatitis have occurred in patients treated with rifabutin. Rifabutin shares with rifampin the property of inducing hepatic microsomal enzymes, with its administration decreasing the half-life of a number of different compounds, including zidovudine, prednisone, digitoxin, quinidine, ketoconazole, propranolol, phenytoin, sulfonylureas, and warfarin. It has less effect than does rifampin on serum levels of indinavir and nelfinavir. Macrolides A discussion of the pharmacology of the macrolides, including their adverse effects and uses in infections other than MAC, is presented in Chapter 47: Antimicrobial Agents: Protein Synthesis Inhibitors and Miscellaneous Antibacterial Agents. Only features of the macrolides related to their use in the treatment of MAC infections are considered here. Antibacterial Activity Clarithromycin is approximately fourfold more active than azithromycin
against MAC bacteria in vitro and is active against most
nontuberculous mycobacteria with the exception of Mycobacterium simiae
at Bacterial Resistance Use of clarithromycin or azithromycin alone in the therapy of MAC infection is associated with the development of resistance after prolonged treatment. For this reason, these drugs should not be used as monotherapy of MAC infection. Therapeutic Uses Clarithromycin (500 mg twice daily) or azithromycin (500 mg daily) is used in combination with ethambutol, with or without rifabutin, for treatment of MAC infection (Shafran et al., 1996; Masur et al., 1993). Treatment should be continued throughout the lifetime of an HIV-infected individual (USPHS, 1999). Untoward Effects With high doses used to treat MAC infections, tinnitus, dizziness, and reversible hearing loss occasionally have occurred. Quinolones A discussion of the pharmacology of the quinolones, including their adverse effects and uses in infections other than MAC, is presented in Chapter 44: Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections. Only features of the quinolones related to their use in the treatment of MAC infections are considered here. Antibacterial Activity Fluoroquinolones, such as levofloxacin, ciprofloxacin, ofloxacin,
fleroxacin, and sparfloxacin, have inhibitory activity against M.
tuberculosis and MAC bacteria in vitro (at concentrations of Bacterial Resistance Single-agent therapy of M. fortuitum infection with ciprofloxacin has been associated with the development of resistance. Therapeutic Uses Ciprofloxacin, 750 mg twice daily or 500 mg three times daily, has been used as part of a four-drug regimen (with clarithromycin, rifabutin, and amikacin) as salvage therapy for MAC infections in HIV-infected patients, with improvement in symptoms (Havlir and Ellner, 2000). Multidrug-resistant tuberculosis has been treated with ofloxacin, 300 or 800 mg each day, in combination with second-line agents. Clofazimine Discussed more fully under drugs for the treatment of leprosy, clofazimine
inhibits most MAC isolates in vitro at levels of 1.6 to 2.0 Amikacin The antibacterial activity and pharmacology of amikacin are discussed
fully in Chapter 46: Antimicrobial Agents: The Aminoglycosides. Amikacin may
have a role as a third or fourth agent in a multiple-drug regimen for MAC
treatment. Most isolates of MAC are inhibited in vitro by 8 to 32 Chemotherapy of Mycobacterium avium Complex Initial pessimism about the treatment of MAC infection has lifted with the availability of clarithromycin and azithromycin. Both of these agents have excellent activity against many strains of MAC, with clinical responses (decrease or elimination of bacteremia, resolution of fever and night sweats) demonstrated even with single-drug therapy. Single-agent therapy, however, has been associated with the emergence of resistant strains. Most clinicians are currently treating MAC infections with clarithromycin or azithromycin plus ethambutol (Haas, 2000; Shafran et al., 1996). In some situations, and with unclear benefits, rifabutin, clofazimine, and/or a quinolone are added to the above regimen. Drug interactions and adverse drug reactions are common with multiple-drug regimens and necessitated drug discontinuation in 46% of patients in one study (Kemper et al., 1992). Clinical improvement should be expected in the first 1 to 2 months of treatment, with sterilization of blood cultures seen as late as 3 months into therapy (Masur et al., 1993). Therapy of MAC infection in HIV-infected individuals should continue for life if the therapy is associated with clinical and microbiological improvement (USPHS, 1999). Isoniazid and pyrazinamide have no role in the treatment of MAC infection. Prophylaxis of MAC infection with clarithromycin or azithromycin should be strongly considered for HIV-infected persons whose CD4 cell count is less than 50/mm3. Clarithromycin and azithromycin are well-tolerated medications that have proven effective at reducing the incidence of MAC infection in this population. With the advent of HAART, it would be a reasonable decision to stop prophylaxis in a patient who responds to anti-HIV therapy with a sustained CD4+ T-lymphocyte count greater than 100/mm3 and a sustained suppression of HIV plasma RNA (USPHS, 1999). |
Drugs for Leprosy
|
Although leprosy (Hansen's disease) is seen
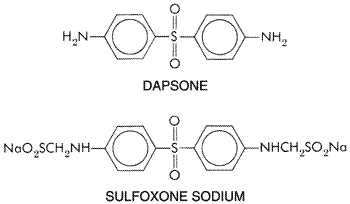
rarely in the Sulfones The sulfones, as a class, are derivatives of 4,4'-diaminodiphenylsulfone (dapsone), all of which have certain pharmacological properties in common. They are discussed here as a class; only dapsone and sulfoxone are considered individually. History The sulfones first attracted interest because of their chemical relationship to the sulfonamides. In the 1940s, sulfones were found to be effective in suppressing experimental infections with the tubercle bacillus and for rat leprosy; this finding was soon followed by successful clinical trials in human leprosy. The sulfones are currently the most important drugs for the treatment of this disorder. Chemistry All the sulfones of clinical value are derivatives of dapsone. Despite the study and development of a large variety of sulfones, this drug remains the agent most useful clinically. The structures of dapsone and sulfoxone sodium are as follows:
Antibacterial Activity Because Mycobacterium leprae does not grow on artificial media, conventional methods cannot be applied to determine its susceptibility to potential therapeutic agents in vitro. Crude sensitivities in vivo can be determined by injecting microorganisms into the foot pads of mice and treating them with the agents to be tested. After 6 to 8 months, the mice are killed, the foot pads are homogenized, and microscopic counts are made of acid-fast microorganisms (Shepard et al., 1976). Dapsone is bacteriostatic, but not bactericidal, for M. leprae, and the estimated sensitivity to the drug is between 1 and 10 ng/ml for microorganisms recovered from untreated patients (Levy and Peters, 1976). M. leprae may become resistant to the drug during therapy. The mechanism of action of the sulfones is the same as that of the sulfonamides. Both possess approximately the same range of antibacterial activity and both are antagonized by paraaminobenzoic acid. Dapsone-resistant strains of M. leprae are termed secondary if they emerge during therapy. Secondary resistance usually is seen in lepromatous (multibacillary) patients treated with a single drug. The incidence is as high as 19% (WHO Expert Committee on Leprosy, 1998). Partial-to-complete primary resistance (seen in previously untreated patients) has been described in from 2.5% to 40% of patients, depending on geographical location (Centers for Disease Control, 1982), although some authorities question the clinical significance of primary resistance (Gelber et al., 1990; Gelber and Rea, 2000). Therapeutic Uses Dapsone is available for oral administration. Several dosage schedules have been recommended (seeTrautman, 1965; Gelber and Rea, 2000). Daily therapy with 100 mg has been successful in adults. Therapy usually is begun with smaller amounts, and doses are increased to those recommended over 1 to 2 months. Therapy should be continued for at least 3 years and may be necessary for the lifetime of the patient. Sulfoxone sodium may be substituted for dapsone in patients in whom the latter drug produces sufficient gastric distress to impede effective therapy. The recommended daily dose of sulfoxone sodium is 330 mg. The use of sulfones in malaria resistant to the usual antimalarial drugs is discussed in Chapter 40: Drugs Used in the Chemotherapy of Protozoal Infections: Malaria. Untoward Effects The reactions induced by various sulfones are very similar. The most common untoward effect is hemolysis of varying degree. This develops in almost every individual treated with 200 to 300 mg of dapsone per day. Doses of 100 mg or less in normal healthy persons and 50 mg or less in healthy individuals with a glucose-6-phosphate dehydrogenase deficiency do not cause hemolysis. Methemoglobinemia also is common, and Heinz-body formation may occur. A genetic deficiency in the NADH-dependent methemoglobin reductase can result in severe methemoglobinemia after administration of dapsone. While diminished red-cell survival usually occurs during the use of sulfones and is presumed to be a dose-related effect of their oxidizing activity, hemolytic anemia is unusual unless the patient also has a disorder of either the erythrocytes or the bone marrow (Pengelly, 1963). The hemolysis may be so severe that manifestations of hypoxia become striking. Anorexia, nausea, and vomiting may follow the oral administration of sulfones. Isolated instances of headache, nervousness, insomnia, blurred vision, paresthesia, reversible peripheral neuropathy (thought to be due to axonal degeneration), drug fever, hematuria, pruritus, psychosis, and a variety of skin rashes have been reported (Rapoport and Guss, 1972). An infectious mononucleosis-like syndrome, which may be fatal, occurs occasionally. The sulfones may induce an exacerbation of lepromatous leprosy by a process thought to be analogous to the JarischHerxheimer reaction. This 'sulfone syndrome' may develop 5 to 6 weeks after initiation of treatment in malnourished people. Its manifestations include fever, malaise, exfoliative dermatitis, jaundice with hepatic necrosis, lymphadenopathy, methemoglobinemia, and anemia. The sulfones may be given safely for many years in doses adequate for the successful therapy of leprosy if proper precautions are observed. Treatment should be initiated with a small dose and the quantity then increased gradually. Patients must be under consistent and prolonged laboratory and clinical supervision. The reactions induced by the sulfones, especially those related to exacerbation of the leprosy, may be very severe and may require the cessation of treatment as well as the institution of specific measures to reduce the threat to life. Absorption, Distribution, and Excretion Dapsone is absorbed rapidly and nearly completely from the
gastrointestinal tract. The disubstituted sulfones, such as sulfoxone, are
absorbed incompletely when administered orally, and large amounts are
excreted in the feces. Peak concentrations of dapsone in plasma are reached
within 2 to 8 hours after administration; the mean half-life of elimination
is about 20 to 30 hours. Twenty-four hours after oral ingestion of 100 mg,
plasma concentrations range from 0.4 to 1.2 The sulfones are distributed throughout total body water and are present in all tissues. They tend to be retained in skin and muscle and especially in liver and kidney; traces of the drug are present in these organs up to 3 weeks after therapy is stopped. The sulfones are retained in the circulation for a long time because of intestinal reabsorption from the bile; periodic interruption of treatment is advisable for this reason. Dapsone is acetylated in the liver, and the rate of acetylation is genetically determined; the same enzyme carries out the acetylation of isoniazid. Daily administration of 50 to 100 mg results in serum levels exceeding the usual minimal inhibitory concentrations, even in rapid acetylators, in whom the serum half-life of dapsone and certain other drugs is shorter than usual. The urinary excretion of sulfones varies with the type of drug; about 70% to 80% of a dose of dapsone is so excreted. The drug is present in urine as an acid-labile mono-N-glucuronide and mono-N-sulfamate in addition to an unknown number of unidentified metabolites. Probenecid decreases the urinary excretion of the acid-labile dapsone metabolites significantly and that of free dapsone to a lesser extent (Goodwin and Sparell, 1969). Rifampin Rifampin has been discussed above with regard to its use in
tuberculosis. This antibiotic is rapidly bactericidal for M. leprae,
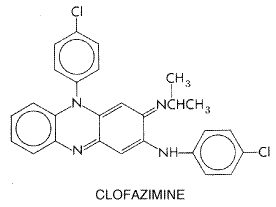
and the minimal inhibitory concentration is less than 1 Clofazimine Clofazimine LAMPRENE) is a phenazine dye with the following structural formula:
Clofazimine appears to preferentially bind to GC-rich mycobacterial DNA (Morrison and Marley, 1976). It is weakly bactericidal against M. intracellulare. The drug also exerts an antiinflammatory effect and prevents the development of erythema nodosum leprosum. Clofazimine is now recommended as a component of multiple-drug therapy for leprosy (see below). The compound also is useful for treatment of chronic skin ulcers (Buruli ulcer) produced by Mycobacterium ulcerans. Clofazimine is absorbed by the oral route and appears to accumulate in tissues. Human leprosy from which dapsone-resistant bacilli have been recovered has been treated with clofazimine with good results. However, unlike dapsone-sensitive microorganisms, in which killing occurs immediately after dapsone is administered, dapsone-resistant strains do not exhibit an appreciable effect until 50 days after initiation of therapy with clofazimine. The daily dose of clofazimine is usually 100 mg (seeLevy et al., 1972). Patients treated with clofazimine may develop red discoloration of the skin, which may be very distressing to light-skinned individuals. Eosinophilic enteritis has also been described as an adverse reaction to the drug (Mason et al., 1977). Miscellaneous Agents Thalidomide seems to be effective for the treatment of erythema nodosum leprosum (Iyer et al., 1971). Doses of 100 to 300 mg per day have been effective. Because of the marked teratogenicity of thalidomide, it should never be administered during pregnancy or to any woman of childbearing age. Ethionamide has been discussed above as an agent for treatment of tuberculosis. It can be used as a substitute for clofazimine in oral doses of 250 to 375 mg per day. New agents that appear promising based on animal trials and limited experience in patients include minocycline, clarithromycin, pefloxacin, and ofloxacin (Gelber and Rea, 2000). Chemotherapy of Leprosy Few physicians other than specialists in the field are called upon to
treat leprosy. Consultation is available with physicians at the National
Hansen's Five clinical types of leprosy are recognized. At one end of the spectrum is tuberculoid leprosy. This form of the disease is characterized by skin macules with clear centers and well-defined margins; these are almost always anesthetic. M. leprae is rarely found in smears made from quiescent lesions but may appear during activity. Virchow cells are not demonstrable. Noncaseating foci with giant cells of the Langhans variety are present. The patient's cell-mediated immune responses are normal, and the lepromin test (intradermal injection of a suspension of heat-killed, bacillus-laden tissue) is invariably positive. The disease is characterized by prolonged remissions with periodic reactivation. At the other end of the spectrum is the widely disseminated lepromatous form of the disease. Patients with this disease have markedly impaired cell-mediated immunity and are frequently anergic; the lepromin test causes no reaction. Lepromatous disease is characterized by diffuse or ill-defined, localized infiltration of the skin, which becomes thickened, glossy, and corrugated; areas of decreased sensation may appear. M. leprae is demonstrable in smears, and granulomas containing bacteria-laden histiocytes (Virchow cells) are present. As the disease progresses, large nerve trunks are involved and anesthesia, atrophy of skin and muscle, absorption of small bones, ulceration, and spontaneous amputations may occur. Three intermediate forms of the disease are recognized: borderline tuberculoid disease, borderline lepromatous disease, and borderline disease (Gelber and Rea, 2000). Patients with tuberculoid leprosy may develop 'reversal reactions,' which are manifestations of delayed hypersensitivity to antigens of M. leprae. Cutaneous ulcerations and deficits of peripheral nerve function may occur. Early therapy with corticosteroids or clofazimine is effective. Reactions in the lepromatous form of the disease (erythema nodosum leprosum) are characterized by the appearance of raised, tender, intracutaneous nodules, severe constitutional symptoms, and high fever. This reaction may be triggered by several conditions but is often associated with therapy. It is thought to be an Arthus-type reaction related to release of microbial antigens in patients harboring large numbers of bacilli. Treatment with clofazimine or thalidomide is effective. The outlook for persons with leprosy has been remarkably altered by successful chemotherapy, surgical procedures that help to restore function and repair disfigurement, and a striking change in the attitude of the public toward patients who have this infection. The social stigma of individuals with this affliction gradually is being replaced by the attitude that considers leprosy a disease caused by a bacterium. Patients with leprosy can be classified as 'infectious' or 'noninfectious' on the basis of the type and duration of disease and effects of therapy. Thus, even 'infectious' patients need not be hospitalized provided that adequate medical supervision and therapy are maintained, the home environment meets specific conditions, and the local health officer concurs in the disposition of the case. Therapy, when effective, heals ulcers and mucosal lesions in months. Cutaneous nodules respond more slowly, and it may take years to eradicate bacteria from mucous membranes, skin, and nerves. The degree of residual pigmentation or depigmentation, atrophy, and scarring depends upon the extent of the initial involvement. Severe ocular lesions show little response to the sulfones. If treatment is initiated before ocular disease is evident, the latter may be prevented. Keratoconjunctivitis and corneal ulceration may be secondary to nerve involvement. The World Health Organization now recommends therapy with multiple drugs for all patients with leprosy (WHO Expert Committee on Leprosy, 1998). The reasons for using combinations of agents include reduction in the development of resistance, the need for adequate therapy when primary resistance already exists, and reduction in the duration of therapy. Dosage recommendations for control programs take a number of practical constraints into account. For patients with large populations of bacteria (multibacillary forms)including lepromatous disease, borderline lepromatous disease, and borderline diseasethe following regimen is suggested: dapsone, 100 mg daily; plus clofazimine, 50 mg daily; plus rifampin, 600 mg and clofazimine (300 mg) once a month under supervision for 1 to 5 years. Some prefer to treat lepromatous leprosy with daily dapsone (100 mg) and daily rifampin (450 to 600 mg) (Jacobson, 1982; Gelber and Rea, 2000). All drugs are given orally. The minimal duration of therapy is 2 years, and treatment should continue until acid-fast bacilli are not detected in lesions. Patients with a small population of bacteria (paucibacillary disease),
including those with tuberculoid, borderline tuberculoid, and indeterminate
disease, should be treated with dapsone, 100 mg daily, plus rifampin, 600 mg
once monthly (under supervision), for a minimum of 6 months. Relapses are
treated by repeating the regimen. A recent clinical trial suggests that
single-dose multidrug therapy with rifampin (600 mg), ofloxacin (400 mg), and
minocycline (100 mg) may be as effective (Single Lesion Multicentre Trial
Group, 1997). More prolonged treatment programs are recommended for patients
in the |
Prospectus
|
Applications of fluoroquinones in the
treatment of mycobacterial disease hold promise. Wider application of
directly observed therapy and the advent of HAART for HIV infection portend a
continued decline in tuberculosis in the |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1866
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved