| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Drugs Used in the Treatment of Asthma
Overview
|
Asthma is an extremely common disorder,
accounting for 1% to 3% of all office visits, 500,000 hospital admissions per
year, and more pediatric hospital admissions than any other single illness.
Annually, more than 5000 children and adults die of asthma attacks in the In this chapter, the data identifying inflammation as the primary
pathophysiological process in asthmatic bronchoconstriction are examined.
Therapy for bronchoconstriction per se, including |
Asthma As an Inflammatory Illness
|
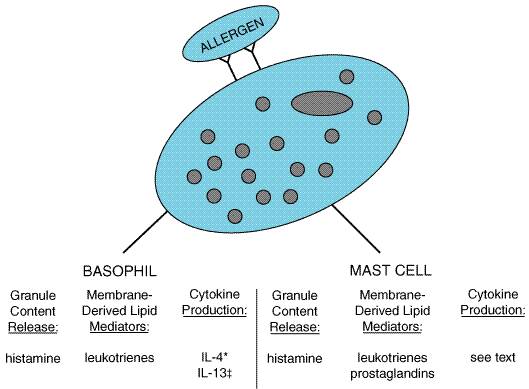
The recognition that asthmatic airway narrowing, both at baseline and during disease exacerbations, is due to inflammation is based on studies involving bronchial lavage and lung biopsies. Increased numbers of inflammatory cells, including eosinophils, basophils, macrophages, and lymphocytes, can be found in bronchoalveolar lavage fluid from asthmatic patients. Even asthmatics with normal baseline lung function and no recent asthma exacerbations have increased numbers of inflammatory cells in their airways. After challenge with allergen, there is a further increase in the numbers of inflammatory cells. Lung biopsies have been performed on normal and asthmatic subjects. Asthmatic subjects have increased airway thickness and an increased number of basophils and other inflammatory cells in lung tissues. The basis for this inflammation is not entirely clear. Most children and adults have clearly defined allergen exposures that are partially or substantially responsible for their asthmatic inflammation. Presumably, these reactions can be at a smoldering level, resulting in continuous mild to moderate inflammation but not overt bronchoconstriction. Epidemiologic studies show that there is a correlation between increasing immunoglobulin E (IgE) levels and prevalence of asthma (Burrows et al., 1989). Figure 281 depicts basophil and mast-cell activation by exposure to allergen. Allergen-specific IgE is bound to the mast cell via Fc receptors. When allergen crosslinks two IgE molecules, basophils and mast cells are activated and release a large number of inflammatory mediators. The mechanisms of this release are now well established and involve dumping of granule contents and synthesis of various lipid mediators (Table 281). Immunological stimulation of basophils also leads to the synthesis of several proinflammatory cytokines, such as interleukin (IL)-4 and IL-13 (Schroeder and MacGlashan, 1997). Which cytokines are generated by mast cells in the airways is not yet clear. The salient feature of this scheme is that an enormous variety of mediators is released, each having more than one potent effect on airway inflammation.
Figure 281. Inflammatory Mediators Released by Activated Mast Cells and Basophils. IL = interleukin *= over 14 hours = over 624 hours; The result of the vasodilation, increased vasopermeability, and an increased display of endothelial leukocytic adhesion molecules is an influx of inflammatory cells from the circulation into the tissues. Lymphocytes, eosinophils, and basophils predominate. Once these newly recruited cells reach the lung, they release their own mediators, which have further inflammatory effects (Table 282). While histamine and leukotriene come from mast cells in an acute reaction, these mediators, together with IL-4 and IL-13, come from basophils in chronic disease. Asthmatic inflammation is characterized by bronchial hyperreactivity and therefore differs from the inflammation found in other conditions, such as pneumonia. The chronic results are airway edema, smooth muscle hypertrophy, epithelial shedding, and bronchial hyperreactivity to nonspecific stimuli such as strong odors, cold air, pollutants, and histamine. Asthmatic airway inflammation may cause increased parasympathetic tone, with resulting bronchial narrowing. The above scheme predicts that a drug affecting only one mediator is unlikely to be of substantial benefit, simply because there are so many mediators participating. For example, histamine clearly is released during allergic asthmatic reactions (Murray et al., 1986), but antihistamines are of little or no benefit in allergic asthma (Holgate, 1994). In contrast, leukotriene inhibitors or an IL-4receptor antagonist have clear effects. One also can predict that drugs that more broadly address asthmatic inflammation (i.e., glucocorticoids) could be of greater therapeutic benefit than agents that address only bronchoconstriction per se. |
Treatment of Asthma
|
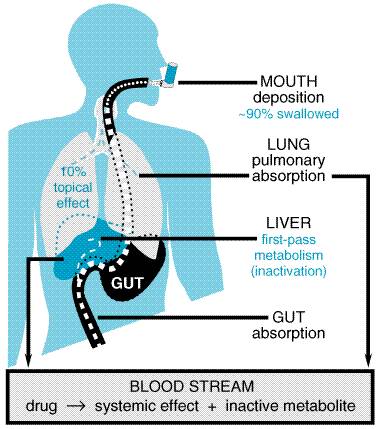
Aerosol Delivery of Drugs Topical application of drugs to the lungs can be accomplished by use
of aerosols. In theory, this approach should produce a high local
concentration in the lungs with a low systemic delivery, thereby
significantly improving the therapeutic ratio by minimizing systemic side
effects. The most commonly used drugs in the treatment of asthma, A review of the chemistry and physics of aerosol delivery systems is
available (Taburet and Schmit, 1994). A schematic diagram of the fate of
therapeutic agents delivered by this route is shown in Figure 282. The
critical determinant of the delivery of any particulate matter to the lungs
is the size of the particles. Particles larger than 10
As
depicted in Figure 282, even under ideal circumstances only a small fraction
of the aerosolized drug is deposited in the lungs, typically 2% to 10%. Most
of the remainder is swallowed. Therefore, to have minimal systemic effects,
an aerosolized drug should be either poorly absorbed from the
gastrointestinal system or rapidly inactivated via first-pass hepatic
metabolism. Furthermore, any maneuvers that result in a higher percentage of
deposition in the lungs and a lower percentage of drug reaching the
gastrointestinal system should improve the therapeutic index. For example,
with metered-dose inhalers, a large-volume 'spacer' can be attached
to the inhaler. A spacer is a tube or expandable bellows that fits between
the inhaler and the patient's mouth; the inhaler discharges into it, and the
patient inhales from it. A spacer can improve markedly the ratio of inhaled
to swallowed drug by limiting the amount of larger particles (>10 The two types of devices used for providing aerosol therapy are metered-dose
inhalers and nebulizers. Both devices provide a range of particle
sizes that includes the desired 1- to 5- An alternative to aerosolized delivery is the use of dry-powder inhalers. These typically use lactose or glucose powders to carry the drugs. One disadvantage of these devices is that a relatively high airflow is needed to suspend properly the powder. Young children, the elderly, and those suffering from a significant asthma exacerbation may not be able to generate such air flow rates. The dry powder can be irritating when inhaled. Storage of dry-powder inhalers in areas where there are wide temperature fluctuations or high humidity can affect their performance.
The history, chemistry, pharmacological properties, and mechanisms of
action of the Mechanism of Action and Use in Asthma The Short-Acting Drugs in this class include albuterol (PROVENTIL VENTOLIN), levalbuterol [XOPENEX, the (R)-enantiomer of albuterol], metaproterenol
(ALUPENT), terbutaline (BRETHAIRE), and pirbuterol (MAXAIR). These drugs are used for acute
inhalation treatment of bronchospasm. Terbutaline (BRETHINE BRICANYL), albuterol, and metaproterenol
also are available in oral dosage form. Each of the inhaled drugs has an
onset of action within 1 to 5 minutes and produces a bronchodilation that
lasts for about 2 to 6 hours. When given in oral dosage forms, the duration
of action is somewhat longer (oral terbutaline, for example, has a duration
of action of 4 to 8 hours). Although there are slight differences in the
relative The mechanism of the antiasthmatic action of short-acting The most effective drugs in relaxing airway smooth muscle and
reversing bronchoconstriction are short-acting Long-Acting Salmeterol xinafoate SEREVENT) is a
long-lasting adrenergic agonist with very high selectivity for the Long-acting There are relatively few studies evaluating the antiinflammatory
effect of adding long-acting Chronic treatment with long-acting Salmeterol should not be used to reverse acute symptoms of asthma.
Rather, physicians prescribing salmeterol also should prescribe a
short-acting Toxicity Owing to their Oral Therapy with The use of orally administered adrenergic agonists for bronchodilation
has not gained wide acceptance, largely because of the greater risk of
producing side effects, especially tremulousness, muscle cramps, cardiac
tachyarrhythmias, and metabolic disturbances (see Chapter 10:
Catecholamines, Sympathomimetic Drugs, and Adrenergic Receptor Antagonists).
There are two situations in which oral Even though stimulation of Glucocorticoids The history, chemistry, pharmacological properties, and mechanisms of action of glucocorticoids are discussed in Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones. Here, the discussion is restricted to their uses in asthma. Barnes and Pedersen (1993) have provided a thorough review of this subject. Systemic glucocorticoid administration long has been employed to treat
severe chronic asthma or severe, acute exacerbations of asthma (McFadden, 1993;
Greenberger, 1992). The development of aerosol formulations significantly
improved the safety of glucocorticoid treatment, allowing it to be used for
moderate asthma (Busse, 1993). Asthmatic subjects who require inhaled Mechanism of Action in Asthma Asthma is a disease associated with airway inflammation, airway hyperreactivity, and acute bronchoconstriction. Glucocorticoids do not relax airway smooth muscle and thus have little effect on acute bronchoconstriction. By contrast, these agents are singularly effective in inhibiting airway inflammation. Very few mechanisms that lead to the inflammatory reaction escape the inhibitory effects of these drugs (Schleimer, 1998). The mechanisms that contribute to the antiinflammatory effect of glucocorticoid therapy in asthma include modulation of cytokine and chemokine production, inhibition of eicosanoid synthesis, marked inhibition of accumulation of basophils, eosinophils, and other leukocytes in lung tissue, and decreased vascular permeability (Schleimer, 1998). The profound and generalized antiinflammatory action of this class of drugs explains why they are the most effective drugs used in the treatment of asthma at present. Inhaled Glucocorticoids Glucocorticoids have long been known to be effective in controlling
asthma, but treatment with systemic glucocorticoids comes with the cost of
considerable unwanted side effects. A major advance in asthma therapy was the
development of glucocorticoids that could be delivered to the lungs via
inhalation. This allowed for the targeting of the drug directly to the
relevant site of inflammation. In so doing, the therapeutic index of the
drugs has been greatly enhanced by substantially diminishing the number and
degree of side effects, without sacrificing clinical efficacy. There are
currently five glucocorticoids available in the Inhaled glucocorticoids are used prophylactically to control asthma, rather than to acutely reverse asthma symptoms. As with all prophylactic therapies, compliance is a significant concern. Issues relating to drug compliance, therefore, become relevant when choosing among the various steroid formulations. Having highly potent glucocorticoid action, the newer drugs can be effective with as little as one or two puffs administered twice or even once daily. This more convenient dosage regimen may be preferred by patients, which in turn translates to better compliance and therefore better asthma control. The appropriate dose of steroid must be determined empirically. Important variables that influence the effective dose include the severity of disease, the particular steroid used, and the device used for drug delivery, as it determines the actual quantity of drug delivered to the lungs (Smaldone, 1997). When determining the optimal dose, it should be kept in mind that maximal improvement in lung function may not occur until after several weeks of treatment. Asthmatic patients maintained on inhaled glucocorticoids show
improvement in symptoms and lowered requirements for 'rescue' with Systemic Glucocorticoids Systemic glucocorticoids are used for acute asthma exacerbations and chronic, severe asthma. Substantial doses of glucocorticoids (e.g., 40 to 60 mg of prednisone daily for 5 days; 1 to 2 mg/kg per day for children) often are used to treat acute exacerbations of asthma (Weinberger, 1987). Although an additional week of therapy at somewhat reduced dosage may be required, the steroids can be withdrawn abruptly once control of the symptoms by other medications has been restored; any suppression of adrenal function appears to dissipate within 1 to 2 weeks. More protracted bouts of severe asthma may require longer treatment and a slow tapering of the dose to avoid exacerbating asthma symptoms and suppressing pituitary/adrenal function. In persistent asthma, alternate-day therapy with oral prednisone was common in the past. Now, most patients considered for this regimen likely can be treated better with high-dose inhaled glucocorticoids. Toxicity Inhaled Glucocorticoids While there is a great deal of enthusiasm for inhaled glucocorticoids
in asthma, local and systemic adverse effects remain a concern (Table 283).
Some portion of any inhaled drug is swallowed. Therefore, inhaled drugs can
reach the circulation by direct absorption from the lung or by absorption
from the gastrointestinal tract. The newer glucocorticoids have extremely low
oral bioavailability due to extensive first-pass metabolism by the liver.
These reach the circulation almost exclusively by absorption from the lung (Brattsand
and Axelsson, 1997). In contrast to the beneficial effects on asthma, which
reach a plateau at about 1600 Systemic Glucocorticoids The adverse effects of systemic administration of adrenocortical steroids are well known (see Chapter 60: Adrenocorticotropic Hormone; Adrenocortical Steroids and Their Synthetic Analogs; Inhibitors of the Synthesis and Actions of Adrenocortical Hormones), but treatment for brief periods (5 to 10 days) causes relatively little dose-related toxicity. The most common adverse effects during a brief course are mood disturbances, increased appetite, loss of glucose control in diabetics, and candidiasis. Leukotriene-Receptor Antagonists and Leukotriene-Synthesis Inhibitors Zafirlukast ACCOLATE) and montelukast (SINGULAIR) are leukotriene-receptor antagonists. Zileuton (ZYFLO) is an inhibitor of 5-lipoxygenase, which catalyzes the formation of leukotrienes from arachidonic acid. History The history of leukotrienes can be traced back to the classical pharmacological studies in the late 1930s by Kellaway and Trethewie (1940). Upon investigating antigen-induced responses in guinea pigs sensitized to egg albumin, they discovered a slow-reacting, smooth-muscle-stimulating substance. They named the substance SRS based on its pharmacological activity and concluded that it was a unique substance found only in immunologically sensitized tissues subsequently challenged with antigen. Decades later, Brocklehurst (1960) renamed SRS as slow-reacting substance of anaphylaxis, or SRS-A. Two pivotal discoveries were required before the importance of SRS-A
in allergic responses was proven. First was the discovery in 1973 by
scientists at Fisons pharmaceutical company of an SRS-A antagonist called FPL
55712 (Augstein et al., 1973), and second was the elucidation by
Samuelsson and colleagues of the structure of SRS-A as a 5-lipoxygenease
product of arachidonic acid, which they termed cysteinyl leukotriene (Murphy
et al., 1979; see Chapter 26: Lipid-Derived Autacoids:
Eicosanoids and Platelet-Activating Factor). Soon thereafter, an enormous
effort was undertaken by the pharmaceutical industry to discover novel
inhibitors of leukotrienes as potential therapeutic agents for asthma. The
strategies taken were either to reduce the synthesis of leukotrienes by
inhibiting the 5-lipoxygenase enzyme, or to antagonize the effects of
leukotrienes at their receptors. This effort bore fruit in the 1990s with the
development of three new drugs now available for the treatment of asthma in
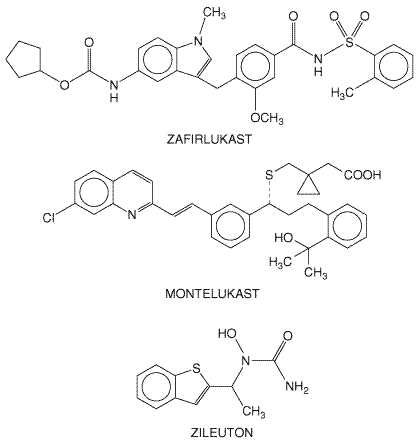
the Chemistry The chemical structures of zafirlukast, montelukast, and zileuton are shown below.
Pharmacokinetics and Metabolism Each of the three leukotriene-modifying drugs is available for oral administration in tablet form. Zafirlukast is rapidly absorbed, with greater than 90% bioavailability. At therapeutic plasma concentrations, it is over 99% protein bound. Zafirlukast is extensively metabolized by the liver cytochrome P450 isozyme CYP2C9. The parent drug is responsible for the drug action with metabolites being less than 10% effective. The half-life of zafirlukast is approximately 10 hours. Montelukast is rapidly absorbed, with about 60% to 70% bioavailability. At therapeutic concentrations, it is highly protein bound (99%). It is extensively metabolized by cytochrome P450 isozymes CYP3A4 and CYP2C9. The half-life of montelukast is between 3 and 6 hours. Zileuton is rapidly absorbed upon oral administration and is extensively metabolized by cytochrome P450 isozymes and by UDP-glucuronosyltransferases. The parent molecule is responsible for the therapeutic action. Zileuton is a short-acting drug, with a half-life of approximately 2.5 hours. Like montelukast, zileuton is highly protein bound (93%). Mechanism of Action in Asthma Leukotriene-modifying drugs act either as competitive antagonists of leukotriene receptors or by inhibiting the synthesis of leukotrienes. The pharmacological properties of leukotrienes are discussed in detail in Chapter 26: Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor. Leukotriene-Receptor Antagonists Cysteinyl leukotrienes (cys-LTs) include LTC4, LTD4, and LTE4. All of
the cys-LTs are potent constrictors of bronchial smooth muscle. On a molar
basis, LTD4 is approximately 1000 times more potent than is histamine as a
bronchoconstrictor (Dahlen et al., 1980). The receptor responsible for
the bronchoconstrictor effect of leukotrienes is the cys-LT1 receptor (Buckner
et al., 1986; Lynch et al., 1999). Although each of the cys-LTs
are agonists at the cys-LT1 receptor, LTE4 is less potent than either LTC4 or
LTD4. Zafirlukast and montelukast are selective, high-affinity, competitive
antagonists of the cys-LT1 receptor (Krell et al., 1990, Jones et al.,
1995). Pranlukast is another cys-LT1receptor antagonist used in some
countries in the treatment of asthma, but it is not approved for use in the The effects of cys-LTs that are potentially relevant to bronchial asthma are not limited to bronchial smooth muscle contraction. Cys-LTs can increase microvascular leakage, increase mucus production, and enhance eosinophil and basophil influx into the airways (Hay et al., 1995). The extent to which inhibiting these non-smooth-muscle effects of leukotrienes contribute to the therapeutic effects of the drugs is not known. It may be noteworthy, however, that zafirlukast significantly inhibits the influx of basophils and lymphocytes entering the airways following experimental allergen challenge in asthmatic subjects (Calhoun et al., 1998). Leukotriene-Synthesis Inhibitors The formation of leukotrienes depends on lipoxygenation of arachidonic acid by the enzyme 5-lipoxygenase. Zileuton is a potent and selective inhibitor of 5-lipoxygenase activity and thus inhibits the formation of all 5-lipoxygenase products. This means that, in addition to inhibiting the formation of the cys-LTs, zileuton inhibits the formation of leukotriene B4 (LTB4), a potent chemotactic autacoid, and other eicosanoids that depend on LTA4 synthesis (see Chapter 26: Lipid-Derived Autacoids: Eicosanoids and Platelet-Activating Factor). In theory, the therapeutic effects of a 5-lipoxygenase inhibitor would include all of those observed with the cys-LT1receptor antagonists, as well as other effects that may result from inhibiting the formation of LTB4 and other 5-lipoxygenase products. The pharmacological actions of cys-LTs are not fully accounted for by activation of the cys-LT1 receptor. For example, cys-LTinduced contraction of vascular smooth muscle (Gorenne et al., 1995) and stimulation of expression of P-selectin by endothelial cells occur via noncys-LT1 receptor subtypes (Pedersen et al., 1997). This provides another theoretical advantage of zileuton over zafirlukast and montelukast, because 5-lipoxygenase inhibitors would inhibit cys-LT effects regardless of the receptor subtypes involved. These theoretical advantages notwithstanding, studies thus far do not support the hypothesis that zileuton is significantly more efficacious than the cys-LT1receptor antagonists in the treatment of asthma. Toxicity There are few adverse effects directly associated with inhibition of leukotriene synthesis or function. This is likely due to the fact that the production of leukotrienes is predominantly limited to sites of inflammation. Zafirlukast and Montelukast In large clinical studies, the adverse effect profile of these drugs was similar to that observed with placebo treatment. In very rare instances, patients taking these drugs developed a systemic eosinophilia and a vasculitis with features similar to Churg-Strauss syndrome. This problem was often associated with the reduction in oral glucocorticoid therapy, and may represent the uncovering of a preexisting disease. Zafirlukast, but not montelukast, may interact with warfarin and increase prothrombin-times, so prothrombin-times should be monitored in those patients subject to this interaction. Zileuton The adverse effect profile in patients taking zileuton is similar to that in patients taking placebo. In about 4% to 5% of patients taking zileuton, however, there is an elevation in liver enzymes. In the majority of patients, this occurs during the first two months of therapy. Zileuton decreases the steady-state clearance of theophylline, resulting in substantive increases in theophylline plasma concentrations. Zileuton also decreases warfarin clearance. Use in Asthma Although leukotriene inhibitors have been proven to be effective
prophylactic treatment for mild asthma, their position in the guidelines for
asthma therapy have not yet been clearly stated. Most clinical studies with
these drugs have been conducted in patients with mild asthma who were not
taking glucocorticoids. In general, the studies show a modest but significant
improvement in lung function and a decrease in symptoms and asthma
exacerbations. In a meta-analysis of clinical trials with zafirlukast, all
studies showed some decrease in the rate of asthma exacerbations, with an
average effect of 50% reduction (Barnes and Miller, 2000). When zafirlukast (Laitinen
et al., 1997) or montelukast (Malmstrom et al., 1999) were
compared with low-dose, inhaled glucocorticoid therapy, the improvement in
lung function and decreased dependence on short-acting More studies are required before the role of these drugs in moderate
and more severe asthma can be adequately assessed. Some clinical trials have
demonstrated an ability of leukotriene antagonists to allow a reduction in
the dose of inhaled steroid needed to control asthma exacerbations (Lofdahl et
al., 1999; Jarvis and Markham, 2000). If this is the case, it may be
particularly relevant in children with more severe asthma. This class of
drugs is not indicated for rapid bronchodilator therapy; thus, patients are
instructed to have appropriate rescue medications available, i.e,
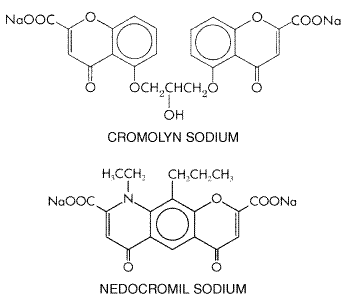
short-acting Cromolyn Sodium and Nedocromil Sodium History and Chemistry Cromolyn
was synthesized in 1965 as part of an attempt to improve on the
bronchodilator activity of khellin, a chromone (benzopyrone) derived
from the plant Ammi visnaga, which had been used by the ancient
Egyptians for its spasmolytic properties (see Shapiro and Knig, 1985).
Although devoid of the bronchodilating capability of the parent compound,
cromolyn was found to inhibit antigen-induced bronchospasm as well as the
release of histamine and other autacoids from sensitized rat mast cells.
Cromolyn has been used in the
Mechanism of Action Cromolyn and nedocromil have been reported to have a variety of activities that may relate to their therapeutic effect in asthma. These include: inhibiting mediator release from bronchial mast cells (Pearce et al., 1989); an ability to reverse increased functional activation in leukocytes obtained from the blood of asthmatic patients (Murphy and Kelly, 1987); suppression of the activating effects of chemotactic peptides on human neutrophils, eosinophils, and monocytes (Kay et al., 1987; Moqbel et al., 1988); inhibition of parasympathetic and cough reflexes (Hargreaves and Benson, 1995; Fuller et al., 1987); and inhibition of leukocyte trafficking in asthmatic airways (Hoshino and Nakamura, 1997). Suffice it to say that the mechanism of action of cromolyn and nedocromil in asthma is not known. Pharmacokinetics For asthma, cromolyn is given by inhalation, using either solutions
(delivered by aerosol spray or nebulizer) or, in some countries but not in
the Toxicity Cromolyn and nedocromil generally are well tolerated by patients. Adverse reactions are infrequent and minor; these include bronchospasm, cough or wheezing, laryngeal edema, joint swelling and pain, angioedema, headache, rash, and nausea. Such reactions have been reported at a frequency of less than 1 in 10,000 patients (see Murphy and Kelly, 1987). Very rare instances of anaphylaxis also have been documented. Nedocromil and cromolyn can cause a bad taste. Use in Asthma The main use of cromolyn (INTAL) and nedocromil (TILADE) is in the treatment of mild to moderate bronchial asthma to prevent asthmatic attacks. These agents are ineffective in treating ongoing bronchoconstriction. When inhaled several times daily, cromolyn will inhibit both the immediate and the late asthmatic responses to antigenic challenge or to exercise. With regular use for more than 2 to 3 months, there is evidence of reduced bronchial hyperreactivity, as measured by response to challenge with histamine or methacholine (see Murphy and Kelly, 1987; Hoag and McFadden, 1991). Nedocromil generally is more effective than cromolyn in animal models and human studies (Brogden and Sorkin, 1993). Nedocromil is approved for use in asthmatic patients 12 years old and older; cromolyn is approved for all ages. Compared to inhaled glucocorticoids, cromolyn and nedocromil are less
potent in controlling asthma. Cromolyn, 2 mg inhaled four times daily, was
not as effective as beclomethasone, 200 The use of cromolyn or nedocromil in addition to inhaled glucocorticoids in moderately severe asthma has been investigated. Several studies have shown that the addition of cromolyn to inhaled glucocorticoid therapy yields no additional benefit (Toogood et al., 1981). Nedocromil may allow a reduction of steroids in patients receiving high doses of inhaled steroids (Brogden and Sorkin, 1993). These studies were short term; whether or not long-term reduction in steroid doses is possible remains to be determined. In one study, the addition of nedocromil, 4 mg four times daily, to high-dose, inhaled glucocorticoid treatment resulted in modest improvements when administered for 8 weeks to patients with moderately severe asthma (Svendsen and Jorgensen, 1991). Because of its limited potency, the use of cromolyn is decreasing. In patients with systemic mastocytosis who have gastrointestinal symptoms due to an excessive number of mast cells in the gastrointestinal mucosa, an oral preparation of cromolyn (GASTROCROM) is effective in reducing symptoms (Horan et al., 1990). The benefits are derived from the topical application rather than systemic absorption; cromolyn is poorly absorbed, and only the gastrointestinal symptoms are improved in the treated patients. Theophylline Theophylline,
a methylxanthine, is among the least expensive drugs used to treat asthma,
and consequently it remains a commonly used drug for this indication in many
countries. In industrialized countries, the advent of inhaled
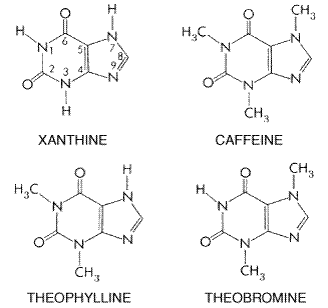
glucocorticoids, Source and History Theophylline, caffeine, and theobromine are three closely related
alkaloids that occur in plants widely distributed geographically. At least
half the population of the world consumes tea (containing caffeine and small
amounts of theophylline and theobromine), prepared from the leaves of Thea
sinensis, a bush native to southern The basis for the popularity of all the caffeine-containing beverages is the ancient belief that they have stimulant and antisoporific actions that elevate mood, decrease fatigue, and increase capacity for work. For example, legend credits the discovery of coffee to a prior of an Arabian convent. Shepherds reported that goats that had eaten the berries of the coffee plant gamboled and frisked about all through the night instead of sleeping. The prior, mindful of the long nights of prayer that he had to endure, instructed the shepherds to pick the berries so that he might make a beverage from them. Classical pharmacological studies, principally of caffeine, during the first half of this century confirmed these experiences and revealed that methylxanthines possess other important pharmacological properties as well. These properties were exploited for a number of years in a variety of therapeutic applications, in many of which caffeine has now been replaced by more effective agents. However, in recent years there has been a resurgence of interest in the natural methylxanthines and their synthetic derivatives, principally as a result of increased knowledge of their cellular basis of action. Chemistry Theophylline, caffeine, and theobromine are methylated xanthines. Xanthine itself is a dioxypurine and is structurally related to uric acid. Caffeine is 1,3,7-trimethylxanthine; theophylline, 1,3-dimethylxanthine; and theobromine, 3,7-dimethylxanthine. The structural formulas of xanthine and the three naturally occurring xanthine derivatives are as follows:
The solubility of the methylxanthines is low and is much enhanced by the formation of complexes (usually 1:1) with a wide variety of compounds. The most notable of these complexes is that between theophylline and ethylenediamine (to form aminophylline). The formation of complex double salts (e.g., caffeine and sodium benzoate) or true salts [e.g., choline theophyllinate (oxtriphylline)] also enhances aqueous solubility. These salts or complexes dissociate to yield the parent methylxanthines when dissolved in aqueous solution and should not be confused with covalently modified derivatives such as dyphylline [1,3-dimethyl-7-(2, 3-dihydroxypropyl)xanthine]. A large number of derivatives of the methylxanthines have been
prepared and examined for their ability to inhibit cyclic nucleotide
phosphodiesterases (Beavo and Reifsnyder, 1990) and to antagonize
receptor-mediated actions of adenosine (Daly, 1982; Linden, 1991), the two
best-characterized cellular actions of the methylxanthines. In general, both
activities are reduced in derivatives that lack substituents at position 1 or
contain substituents at position 7, as compared with the corresponding
1,3-dialkylxanthine. For example, the order of potency for the naturally
occurring methylxanthines is theophylline > caffeine > theobromine.
Congeners of theophylline with larger nonpolar substituents at positions 1
and 3 usually display enhancement of both activities (Choi et al.,
1988). Addition of aromatic, cyclohexyl, or cyclopentyl groups at position 8
usually markedly increases affinity for adenosine receptors but reduces
inhibition of cyclic nucleotide phosphodiesterases (Martinson et al.,
1987). Although neither caffeine nor theophylline discriminates among the
subtypes of adenosine receptors (see below), certain 8-substituted
derivatives of 1, 3-dipropylxanthine display marked selectivity for A1
receptors, while some analogs of caffeine display appreciable selectivity for
A2 receptors. In addition, certain tricyclic nonxanthine compounds
are potent antagonists at adenosine receptors ( Mechanism of Action Theophylline inhibits cyclic nucleotide phosphodiesterase enzymes (PDEs). PDEs catalyze the breakdown of cyclic AMP and cyclic GMP to 5'-AMP and 5'-GMP, respectively. Inhibition of PDEs will lead to an accumulation of cyclic AMP and cyclic GMP, thereby increasing the signal transduction through these pathways. It is now recognized that cyclic nucleotide PDEs are members of a superfamily of at least eleven families of genetically distinct enzymes (Soderling and Beavo, 2000). Theophylline and related methylxanthines are relatively nonselective in the PDE subtypes they inhibit. The potency and efficacy of PDE inhibitors in affecting cell function is dependent on the basal level of cyclic nucleotide production. Cyclic AMP and cyclic GMP production in cells is regulated by endogenous receptor-ligand interactions leading to activation of adenylyl cyclase and guanylyl cyclase, respectively (see Chapter 2: Pharmacodynamics: Mechanisms of Drug Action and the Relationship Between Drug Concentration and Effect). Diffusable mediators such as nitric oxide and related molecules also may lead to increases in cyclic GMP by direct interaction with guanylyl cyclase. Inhibitors of PDEs therefore can be thought of as drugs that enhance the activity of endogenous autacoids, hormones, and neurotransmitters that signal via cyclic nucleotide messengers. This may explain why the in vivo potency often is increased relative to that observed in vitro. Theophylline also is a competitive antagonist at adenosine receptors (Fredholm and Persson, 1982). Adenosine can act as an autacoid and transmitter with myriad biological actions. Of particular relevance to asthma are the observations that adenosine can cause bronchoconstriction in asthmatics and potentiate immunologically induced mediator release from human lung mast cells (Cushley et al., 1984; Peachell et al., 1988). Inhibition of the actions of adenosine must therefore also be considered when attempting to explain the mechanism of action of theophylline (Feoktistov et al., 1998). Pulmonary System Theophylline effectively relaxes airway smooth muscle and thus can be classified as a bronchodilator. This likely contributes to the acute therapeutic efficacy in asthma. Evidence supports a role for both adenosine receptor antagonism and PDE inhibition in the bronchodilating effect of theophylline. Adenosine does not directly contract human isolated bronchial smooth muscle, but when inhaled acts as a potent bronchoconstrictor in asthmatic subjects (Cushley et al., 1984). Therefore, inhibition of this function of adenosine may contribute to theophylline-induced bronchodilation in some asthmatic subjects. Inhibition of PDE isozymes type III and IV effectively relaxes human isolated bronchial smooth muscle (Torphy et al., 1993). It thus seems likely that inhibition of PDEs also contributes to the bronchodilating effect of theophylline. Also seeming to support a role for PDE inhibition in the mechanism of bronchodilator action of theophylline have been studies with a related methylxanthine drug, enprofylline (3-propylxanthine), which has been extensively studied in Europe for use in the treatment of asthma. Enprofylline is more potent than theophylline as a bronchodilator, but is much less potent than theophylline as an antagonist at most types of adenosine receptors (Pauwels et al., 1985). The latter point, however, needs to be interpreted cautiously. Activation of the A2B subtype of adenosine receptor causes several proinflammatory effects, and both theophylline and enprofylline are potent competitive antagonists of A2B adenosine receptors (Feoktistov et al., 1998). Theophylline also inhibits synthesis and secretion of inflammatory mediators from numerous cell types including mast cells and basophils (Page, 1999). This effect of theophylline likely is due to PDE inhibition and can be mimicked in large part with drugs that selectively inhibit the PDE IV isozymes (Torphy and Undem, 1991). It has been argued that, at therapeutic concentrations, the antiinflammatory effect of theophylline may be more relevant to the drug's therapeutic actions than is direct bronchodilation, but this remains unproven (Page, 1999). A discussion of pharmacological properties of theophylline and other methylxanthines involving other organ systems can be found in previous editions of this book. Absorption, Fate, and Excretion The methylxanthines are absorbed readily after oral, rectal, or parenteral administration. Absorption from rectal suppositories is slow and unreliable. Theophylline administered in liquids or uncoated tablets is rapidly and completely absorbed. Absorption also is complete from some, but not all, sustained-release formulations (see Hendeles and Weinberger, 1982). In the absence of food, solutions or uncoated tablets of theophylline produce maximal concentrations in plasma within 2 hours; caffeine is more rapidly absorbed, and maximal plasma concentrations are achieved within 1 hour. Numerous sustained-release preparations of theophylline are available, designed for dosing intervals of 8, 12, or 24 hours. These preparations cause marked interpatient variability with regard to the rate and extent of absorption and especially the effect of food and time of administration on these parameters (see Symposium, 1986a). Thus, it has become necessary to calibrate a given preparation in a given patient and to avoid substituting one apparently similar product for another. Food ordinarily slows the rate of absorption of theophylline but does not limit its extent. With sustained-release preparations, food may decrease the bioavailability of theophylline within some products but may increase it with others. High-carbohydrate, low-protein diets decrease theophylline elimination, whereas low-carbohydrate, high-protein diets and consumption of 'char-broiled' meat increase elimination. Recumbency or sleep also may reduce the rate or extent of absorption to an important degree. These factors make it difficult to maintain relatively constant concentrations of theophylline in plasma throughout the day. Fortunately, it also has become apparent that the concentrations required to alleviate asthmatic symptoms do not remain constant, and the emphasis has shifted toward designing dosing regimens that ensure peak concentrations in the early morning hours, when symptoms frequently worsen (see Symposium, 1988a). Methylxanthines are distributed into all body compartments; they cross the placenta and pass into breast milk. The apparent volumes of distribution for caffeine and theophylline are similar and usually are between 0.4 and 0.6 liter/kg. These values are considerably higher in premature infants. Theophylline is bound to plasma proteins to a greater extent than is caffeine, and the fraction bound declines as the concentration of methylxanthine increases. At therapeutic concentrations, the protein binding of theophylline averages about 60%, but it is decreased to about 40% in newborn infants and in adults with hepatic cirrhosis (see Hendeles and Weinberger, 1982). Methylxanthines are eliminated primarily by metabolism in the liver. Less than 15% and 5% of administered theophylline and caffeine, respectively, are recovered in the urine unchanged. Caffeine has a half-life in plasma of 3 to 7 hours; this increases by about twofold in women during the later stages of pregnancy or with long-term use of oral contraceptive steroids. In premature infants, the rate of elimination of both methylxanthines is quite slow. The average half-life for caffeine is more than 50 hours, while the mean values for theophylline obtained in various studies range between 20 and 36 hours. However, the latter values include the extensive conversion of theophylline to caffeine in these infants (see Symposium, 1981; Roberts, 1984). There is marked interindividual variation in the rate of elimination of theophylline, due to both genetic and environmental factors; fourfold differences are not uncommon (see Lesko, in Symposium, 1986a). The half-life averages about 3.5 hours in young children, while values of 8 or 9 hours are more typical of adults. In most patients the drug obeys first-order elimination kinetics within the therapeutic range. However, at higher concentrations zero-order kinetics becomes evident because of saturation of metabolic enzymes. This prolongs the decline of theophylline concentrations to nontoxic levels. The disposition of methylxanthines also is influenced by the presence of other agents or of disease (see Jonkman, in Symposium, 1986a). For example, the clearance of theophylline is increased nearly twofold during the administration of phenytoin or barbiturates; cigarette smoking or the administration of rifampin or oral contraceptives produces smaller but appreciable increases in theophylline clearance. By contrast, the administration of cimetidine or certain macrolide antibiotics (e.g., erythromycin) reduces the clearance of theophylline. Although there have been reports to the contrary, neither glucocorticoids nor immunization with purified subvirion influenza vaccine appear to have a significant effect, although acute viral infections and interferon can reduce theophylline clearance. The half-life of theophylline can be quite prolonged in patients with hepatic cirrhosis, congestive heart failure, or acute pulmonary congestion, and values of more than 60 hours have been observed. Although scarcely detectable in adults, the conversion of theophylline to caffeine is an important metabolic pathway in preterm infants (see Symposium, 1981; Roberts, 1984). Caffeine accumulates in plasma to a concentration approximately 25% that of theophylline and is one of the urinary products. About 50% of the theophylline administered to such infants appears in the urine unchanged; the excretion of 1,3-dimethyluric acid, 1-methyluric acid, and caffeine accounts for nearly all of the remainder. Toxicology Fatal intoxications with theophylline have been much more frequent
than with caffeine. Rapid intravenous administration of therapeutic doses of aminophylline
(500 mg) sometimes results in sudden death that is probably due to cardiac
arrhythmias, and the drug should be injected slowly over 20 to 40 minutes to
avoid severe toxic symptoms. These include headache, palpitation, dizziness,
nausea, hypotension, and precordial pain. Additional symptoms of toxicity are
tachycardia, severe restlessness, agitation, and emesis; these effects are
associated with plasma concentrations of more than 20 Most toxicity is the result of repeated administration of theophylline
by either oral or parenteral routes. Although convulsions and death have
occurred at plasma concentrations as low as 25 The widespread use of sustained-release preparations of theophylline
has renewed emphasis on measures to prevent continued absorption,
particularly the use of oral activated charcoal and of sorbitol as a
cathartic (Goldberg et al., 1987); multiple doses of oral charcoal
also will accelerate clearance of theophylline. However, when plasma
concentrations exceed 100 Behavioral Toxicity As noted above, moderate doses of caffeine can provoke intense feelings of anxiety, fear, or panic in some individuals. Even subjects with a history of light-to-moderate use of caffeine experience tension, anxiety, and dysphoria after ingesting 400 mg or more of the drug (see Griffiths and Woodson, in Symposium, 1988b). In infants who have received treatment for apnea of prematurity, theophylline may produce persistent changes in sleep-wake patterns (Thoman et al., 1985), but long-term effects on behavior or cognitive development have yet to be identified (see Aranda et al., in Symposium, 1986a). There has been mounting concern that the treatment of asthmatic children with theophylline might produce depression, hyperactivity, or other behavioral toxicity. However, a study of academic performance of asthmatic children treated or not with theophylline showed equal academic performance in asthmatic and nonasthmatic children (Lindgren et al., 1992). Even though it is difficult to factor out specific effects of theophylline from those caused by the illness or by other features of the treatment regimen, many investigators believe that most children will benefit from the use of alternative means of controlling their symptoms. Use in Asthma Theophylline has proven efficacy as a bronchodilator in asthma and formerly was considered first-line therapy. It now has been relegated to a far less prominent role, primarily because of the modest benefits it affords, its narrow therapeutic window, and the required monitoring of drug levels (Stoloff, 1994; Nasser and Rees, 1993). Nocturnal asthma can be improved with slow-release theophylline preparations (Self et al., 1992), but other interventions such as inhaled glucocorticoids or salmeterol are probably more effective (Meltzer et al., 1992). Some pediatricians favor theophylline over inhaled glucocorticoids because of the theoretic potential for growth suppression. However, in most circumstances, mild or moderate asthma that can be controlled with theophylline likely can be controlled with cromolyn or nedocromil, thus avoiding potential glucocorticoid side effects. There are few data to support the routine use of theophylline in the treatment of acute, severe bronchospasm (Fanta et al., 1986; Rossing et al., 1980). Some chronic asthmatic patients benefit from control of nocturnal symptoms with slow-release theophylline preparations. Therapy is usually initiated by the administration of 12 to 16 mg/kg
per day of theophylline (calculated as the free base) up to a maximum of 400
mg per day for at least 3 days (Weinberger, 1987). Children <1 year old
require considerably less; the dose in mg/kg per day may be calculated as 0.2
X (age in weeks) + 5.0. Starting with these low doses minimizes the early
side effects of nausea, vomiting, nervousness, and insomnia, which often
subside with continued therapy, and virtually eliminates the possibility of
exceeding concentrations of 20 Apnea of Preterm Infants Episodes of prolonged apnea, lasting more than 15 seconds and
accompanied by bradycardia, are not infrequent occurrences in premature
infants. They pose the threat of recurrent hypoxemia and neurologic damage.
Although they often are associated with serious systemic illness, no specific
cause is found in many instances. Beginning with the work of Kuzemko and
Paala (1973), methylxanthines have undergone numerous clinical trials for the
treatment of apnea of undetermined origin. The oral or intravenous
administration of methylxanthines can eliminate episodes of apnea that last
more than 20 seconds, and markedly reduces the number of episodes of shorter
duration (see Symposium, 1981; Roberts, 1984; Aranda et al., in
Symposium, 1986a). Satisfactory responses may occur with plasma
concentrations of theophylline of 4 to 8 Although effects on the growth or development of infants following treatment with methylxanthines have not been detected, the evidence is far from definitive. Therapy is thus continued for as brief a period as possible, usually only a few weeks. Anticholinergic Agents There is a long history of the use of anticholinergic agents in the
treatment of asthma. These agents are discussed in detail in Chapter 7:
Muscarinic Receptor Agonists and Antagonists. With the advent of inhaled The bronchodilation produced by ipratropium in asthmatic subjects develops more slowly and is usually less intense than that produced by adrenergic agonists. Some asthmatic patients may experience a useful response lasting up to 6 hours. The variability in the response of asthmatic subjects to ipratropium presumably reflects differences in the strength of parasympathetic tone and in the degree to which reflex activation of cholinergic pathways participates in generating symptoms in individual patients. Hence, the utility of ipratropium must be assessed on an individual basis by a therapeutic trial. The pharmacological properties and therapeutic uses of ipratropium have been reviewed by Gross (1988) (see also Symposium, 1986b). Combined treatment with ipratropium and Drug Therapy of Asthma in Special Circumstances Pediatric Asthma The pathophysiology of asthma in children appears similar to that in adults (Hill et al., 1992). International guidelines (Rachelefsky and Warner, 1993) and thorough reviews (Van Bever and Stevens, 1992; Moffitt et al., 1994) dealing with the treatment of asthma in children have been published. In general, treatment strategies for children do not differ substantively from those for adults, except that more emphasis is placed on a trial of antileukotriene therapy, nedocromil (age 12 and greater), or cromolyn (Van Bever and Stevens, 1992) to avoid possible complications from glucocorticoids. Although inhaled glucocorticoids may impair growth velocity, a large meta-analysis found that final adult height appears to be unaffected by use of these agents (Allen et al., 1994). Indeed, good control of asthma probably is important in allowing good growth, since poorly controlled asthma itself inhibits growth. Use of oral prednisone in asthma is associated with slightly diminished growth, in terms of attaining final predicted height (Allen et al., 1994). Metered dose inhalers require substantial dexterity and cannot be used by children younger than 5 years of age. This limitation dictates use of either nebulized solutions or parenteral therapy in this patient population. Emergency-Room Patients
Hospitalized Patients In addition to regular use of inhaled Asthma exacerbations requiring hospitalization are handled essentially no differently in children than in adults; treatment with systemic glucocorticoids is required. The dose recommended is 1 to 2 mg/kg per day, divided into four doses. The once-common practice of instituting continuous isoproterenol infusions in children with asthma exacerbations has not been proven to be effective. Maguire et al. (1986) showed that such infusions in children are associated with detectable levels of cardiac-specific creatinine kinase in serum. These infusions also can be associated with tachyarrythmias. At present there is little to recommend such infusions. Asthma during Pregnancy and Lactation Poorly controlled asthma can adversely affect the outcome of pregnancy
and even cause maternal or fetal death. Asthma affects up to 5% of pregnant
women. In the past, asthma frequently caused significant difficulty during
pregnancies. With the recognition by patient and physician of the need for
excellent preventive control of asthma during pregnancy, complications of
pregnancy by asthma should be rare. A consensus conference published its
recommendations concerning the treatment of asthma during pregnancy (NIH,
1993). In general, essentially the same guidelines should be used for
treating pregnant asthmatic patients as for treating nonpregnant asthmatic
patients. Although most drugs used to treat asthma are FDA category C (not
proven to be safe for use during pregnancy), some are in category B
(cromolyn, nedocromil, terbutaline, leukotriene modifiers), and there is a
large clinical experience with inhaled Except for a few studies in animals in which high systemic drug doses
were used, there is no evidence that Antiinflammatory treatment to prevent asthma exacerbations is
indicated whenever pregnant asthmatic patients require daily inhaled Despite a long history of successful use of theophylline preparations
in pregnancy, this drug is now infrequently used, in part because of its limited
effectiveness and narrow therapeutic window. Theophylline elimination is
affected by pregnancy, but to a variable degree. The increased glomerular
filtration rate associated with pregnancy increases the rate of elimination
of theophylline; conversely, metabolic elimination of theophylline by the
liver is decreased. In the last trimester of pregnancy, the overall effect is
an approximately 30% diminished rate of elimination of theophylline. Because
of marked interindividual variability and the changes associated with
progression of pregnancy, frequent drug level monitoring is required. When
maternal levels exceed 20 Use of Asthma Drugs in Rhinitis Seasonal allergic rhinitishay feveris caused by deposition of
allergens on the nasal mucosa, resulting in an immediate hypersensitivity
reaction. This reaction usually is not accompanied by asthma, because the
allergens usually are contained in particles too large to be inhaled into the
lower airways (e.g., pollens). Treatment for allergic rhinitis is
similar to that for asthma. Topical glucocorticoids (beclomethasone, mometasone,
budesonide, flunisolide, fluticasone, triamcinolone acetonide) or cromolyn
can be highly effective with minimal side effects, particularly if treatment
is instituted immediately prior to the allergy season. Topical
glucocorticoids can be administered twice daily (beclomethasone, flunisolide)
or even once daily (budesonide, mometasone, fluticasone, triamcinolone).
Cromolyn usually requires dosing three to six times daily for full effects.
Rare instances of local candidiasis have been reported with glucocorticoids
and probably can be avoided by rinsing the mouth after use. Unlike in asthma,
antihistamines (Chapter 25: Histamine, Bradykinin, and Their Antagonists)
afford considerable, though incomplete, symptom relief in allergic rhinitis.
Nasal decongestants rely on Perennial allergic rhinitis, caused by exposure to allergens present year-round, such as dust mites or animal dander, can be treated similarly. However, since this situation requires continuous exposure to medicines such as topical glucocorticoids, alternatives such as modifying the patient's environment and using immunotherapy (allergen desensitization) should be considered. Use of Asthma Drugs in Chronic Obstructive Pulmonary Disease Emphysema can be prevented or its progression slowed by the patient's ceasing to smoke (Ferguson and Cherniack, 1993). Pharmacological interventions can help patients to stop smoking. Nicotine gum and transdermal patches are moderately useful when combined with other interventions such as support groups and physician encouragement. Clonidine may be helpful in reducing the craving for cigarettes. Treatment of nicotine addiction is discussed in Chapter 24: Drug Addiction and Drug Abuse. The pharmacological treatment of established emphysema resembles that
of asthma largely because the inflammatory/bronchospastic component of a
patient's disease is the aspect amenable to therapy (Ferguson and Cherniack,
1993). For patients with emphysema who have a significant degree of active
inflammation with bronchospasm and excessive mucus production, symptomatic
use of inhaled ipratropium or a In fact, there are many patients who have nearly pure emphysema,
without a significant degree of reversible inflammation or
bronchoconstriction. Nevertheless, these patients often receive prolonged
courses of ipratropium,
In a minority of patients, emphysema results from a genetic deficiency
of the plasma proteinase inhibitor Recombinant DNAse (dornase alfa, PULMOZYME) is available as a nebulizer solution for treatment of cystic fibrosis. In cystic fibrosis, inspissated secretions containing large numbers of inflammatory cells lodge in the smaller airways, causing obstruction. A substantial portion of the viscosity of the purulent material is due to the DNA from the nuclei of lysed cells. Inhaled DNAse has been shown to aid in clearing these secretions and improving pulmonary function in patients with cystic fibrosis (Harris and Wilmott, 1994; Wilmott and Fiedler, 1994). Efficacy trials are currently underway to assess DNAse treatment in adult COPD exacerbations, where purulent bronchial secretions also contribute to airway obstruction. |
Prospectus
|
Over the past decade, the hypothesis that there may be a more selective and thus safer approach to controlling airway inflammation than glucocorticoid therapy has motivated much of the drug discovery effort in asthma research. In various stages of development are agents aimed at inhibiting certain cytokines, chemokines, or adhesion molecules and thereby selectively decreasing the influx of eosinophils and other leukocytes into the airways. Other approaches have targeted the atopic reaction for selective intervention. Monoclonal antibodies aimed at inhibiting the binding of immunoglobulin E (IgE) to receptors on mast cells and basophils are in clinical trials for asthma and other allergic diseases. These antibodies, in theory, would inhibit the immediate hypersensitivity reaction at its earliest stage, thus preventing allergic inflammation in the airways. Neuropeptide receptor antagonists, particularly antagonists at neurokinin receptors, are under development to inhibit neurogenic components of inflammation. These agents also may inhibit reflex activity in the airways. Another strategy to quell the inflammation associated with asthma and chronic obstructive pulmonary disease is to develop drugs that are isozyme-selective phosphodiesterase (PDE) inhibitors. The finding that the predominant PDEs in inflammatory cells are members of the PDE-IV family has led to the development of drugs that are PDE-IVselective inhibitors. This type of drug may provide antiinflammatory actions without many of the side effects associated with nonselective PDE inhibitors such as theophylline. For further discussion of asthma, see Chapter 236 in Harrison's
Principles of Internal Medicine, 16th ed., Acknowledgment The authors wish to acknowledge Theodore W. Rall and William E. Serafin, the authors of this chapter in the eighth and ninth editions of Goodman and Gilman's The Pharmacological Basis of Therapeutics, respectively, some of whose text has been retained in this edition. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 3477
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved