| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
PROTEOMICS: BIOMEDICAL AND PHARMACEUTICAL APPLICATIONS
A constancy in human history is that new discoveries have systematically been used to improve health and increase the duration and quality of life. From the use of flints to perform primitive surgery during prehistory, to the preparation of plant extracts to cure diseases throughout the world and historic times, the will to turn new basic knowledge into practical tools for medicine is probably a shared value of all civilizations and cultures. During the first part the 20th century, shortly after discovering the radioactive
compounds,
radium and polonium, the physicist Marie Curie created in
science can have a rapid and significant impact in medicine. The second half of the last century was intellectually dominated by the discovery of the genetic code and the description of the structure of its carrying molecule, the DNA, with the end of this era essentially marked by the complete sequencing of the human genome. This major achievement was based on considerable technological progress in biochemistry and bioinformatics, but a significant impetus was the promise of a better understanding of human health through the discovery of genes involved in pathology. However, during the course of human genome sequencing, the scientific community and the public progressively realized that the accomplishment of this huge project was not going to revolutionize, on its own, the field of biomedicine. In large part, this was due to the presence of a significant number of genes for which function was (and remains) unknown, and because biology is much more complex than the structure and functioning of nucleic acids. In a way, the genome project somehow contributed, for a while, to shorten our vision of biology.
The beginning of the 21st century has seen the rediscovery of the complexity of life at the molecular and cellular levels. While genes are the support for transmission of genetic information, most of the chemical reactions inside cells are carried out by proteins, which are the functional products of gene expression. This has given way to the realization that proteomics is the most relevant way to identify markers of pathologies and therapeutic targets. Whereas it is now clear that the number of human genes is around 35,000, there is no consensus on the number of proteins. Taking under consideration alternative splicing of mRNA and post-translational modifications, most of the current projections of protein number range from 5 X to 5 X , but interestingly it cannot be excluded that the number of proteins might in fact be closer to infinity. To add further complexity, proteins interact one with another and the proteome is intrinsically dynamic as it varies between cell types and from physiological condition to condition as occurs in living cells. There is no doubt that defining the human proteome is going to be a much more difficult task than the sequencing of genome.
Together, it might not be an exaggeration to say that the major outcome of the human genome sequencing has finally been to open the way to the exploration of the proteome, transferring the goals and hopes in terms of biomedical and pharmaceutical applications. This is clearly a challenging, but also a promising heritage that this book explores through a series of ongoing experiences and projects representative of the new era in which biology and medicine have now entered.
Proteomics Today, Proteomics Tomorrow
. INTRODUCTION
The basic mantra DNA makes RNA makes protein is the cornerstone of molecular and cellular biology and has interestingly enough now devolved into three sub-sciences: genomics, transcriptomics and proteomics. Each field is driven by its own technologies and each offers its own potential in providing new knowledge that will be useful in diagnosing and treating human and animal diseases. However, the boundaries separating these fields are diffuse and indeed there are no exact definitions for any of them. This is particularly true of proteomics, which clearly encompasses the greatest territory, because there are probably as many views of what proteomics encompasses (and therefore expectations) as there are interested parties (Huber, 2003).
In general, genomics is the study of the genome of an organism, which includes identifying all the genes and regulatory elements, and de facto the sequence of all of the DNA making up the genetic material. As the number of determined genomes rises (it is presently around 50), comparative analyses also become valuable. The expression of the genome leads to the formation of the transcriptome, which is the compliment of mRNAs that reflects the structural genes of an organism; however, this is more complex primarily due to the fact that the transcription of the genome is sensitive to a variety of factors related to the context of the cell and its environment. Indeed, at any given moment, only a subset of the full set of genes will be expressed and this is true whether it is a single cell organism or a cell from the organ of a higher eukaryote. As conditions change the expressed transcriptome will change too. Thus, the major confounding factor is time, a dimension that is largely absent in genomic studies. In eukaryotes, splicing events can produce several mRNAs from a single gene, which introduces yet another element of diversity. Clearly, transcriptomics (the description of the transcriptome and its expression) is potentially far more complex than genomics.
The conversion of the transcriptome to its protein products produces the corresponding proteome. This term was introduced by Marc Wilkins to describe all proteins expressed by a genome, cell or tissue in work for his doctoral degree (Cohen, 2001, Huber, 2003) and has been essentially universally adopted. In parallel with genomics and transcriptomics, proteomics is thus the description of a proteome. Since in cells and whole organisms the transcriptome is temporally dependent, it follows that the corresponding proteome is as well. However, the proteome is distinguished from both the genome and transcriptome in another very important way: whereas in the first two the information is essentially linear (sequential), the proteome is defined by both sequential and three-dimensional structures. Along with the many co-/posttranslational modifications that also occur, the complexity of the proteome is clearly much, much greater and presents a problem that is of undefined magnitude. In fact, it is this realization that really distinguishes proteomics from either genomics or transcriptomics.
While both the genome and transcriptome (albeit that this has yet to be achieved in the latter case) can be seen as finite and thus definable, even for a entity as large as the human genome, the description of the complete proteome of a single cell or even a simple organism, let alone higher eukaryotes, is not presently imaginable and may well not be achievable, i.e. it may be effectively infinite in size (Cohen, 2001, Huber, 2003).
It must be noted that everyone does not accept this position and where the term proteome is applied to the third potential source included by Wilkins, i.e. tissue, in contradistinction to genome (organism) or cell, something approaching a complete description might be possible. The plasma proteome is a case in point (see below). In the ensuing remarks, a more detailed definition of proteomics is developed along with an outline of how this has produced a working agenda for todays proteomic studies.
Illustrations of some of what this has yielded and is likely to yield in the foreseeable future - tomorrows proteomics - will be contrasted with some of the more unlikely promises that have been made.
PROTEOMICS - A UNIVERSAL DEFINITION
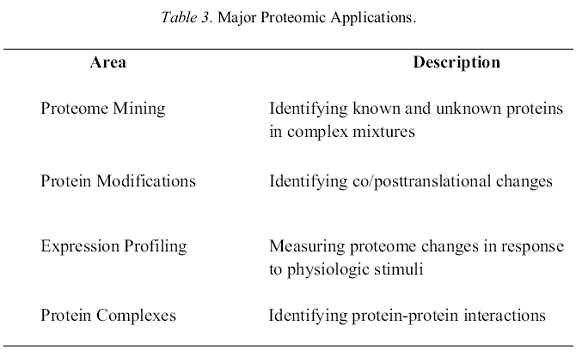
Table 1 provides a four-part definition for proteomics that outlines the scope and nature of the field and thus the information required to fully describe a proteome. Each part represents a different aspect of proteomic research and some have even acquired names for all or part of what they encompass. A brief description of these areas (and sub areas) follows.
Table 1. Proteomics - A Universal Definition.
Determination of the structure and function of the complete set of proteins produced by the genome of an organism, including co- and posttranslational modifications
Determination of the all of the interactions of these proteins with small and large molecules of all types
Determination of the expression of these proteins as a function of time and physiological condition
The coordination of this information into a unified and consistent description of the organism at the cellular, organ and whole animal levels

The first part deals primarily with the identification of all the protein components that make up the proteome and can be viewed in some respects as a giant catalog. The simplest form of this list would be the one to one match up of proteins with genes. This can be most easily accomplished in prokaryotes where mRNA splicing does not occur, but still is a realizable, if much larger and more elusive, goal in eukaryotes. It is this aspect of proteomics that most closely parallels genomics and transcriptomics and what undoubtedly gives workers from those fields the highly questionable sense that the proteome is a finite and therefore determinable entity. The match up, however, extends beyond the sequence level as one must also add to the catalog the three dimensional structure of each protein and all covalent modifications that accompany the formation and maturation of that protein, beginning with the processing of the N-terminus and ending with its ultimate proteolytic demise. This is a considerably larger challenge and one that has not been achieved for any proteome and likely will not be for some time.
Indeed, the structural aspects of the proteome, which have also been referred to as structural genomics and structural proteomics (the difference between these terms is obscure at best), lag well behind the identification of sequences and covalent modifications (Sali et al.
The final component of this part of the definition is the determination of function for each protein and it is here that one must begin to seriously depart from the cataloging concept. While one can usually connect one or more functions (catalytic, recognitive, structural, etc.) to most proteins, ascribing single functions to proteins is at best a gross simplification of the situation. As taken up in part three, protein-protein interactions, with their resultant effects on activity, are enormously widespread and since the presence or absence of these interactions, along with many co-/posttranslational modifications are time (expression) dependent, simple listings are at best a draconian description of a proteome and are really inadequate to describe function in the cellular context.
While the concept of a protein catalog undoubtedly existed, at least in the minds of some people, prior to the coining of the term proteome, the realization that protein-protein interactions were a dominant feature of proteome function was certainly considerably less clear. However, such is the case and the second part of the definition requires a detailed knowledge of all of these recognitive events, both stable and transient. The elucidation of these interactions and the underlying networks that they describe have been termed cell-mapping proteomics (Blackstock & Weir, 1999). This in fact has been a quite productive area of proteomic research, as described below.
The third part of the definition clearly represents the greatest challenge in terms of data collection. If the first two parts can be said to loosely represent the who, what and how of proteomics, then the third part is the when and where, and is by far the most dynamic of the three. Obtaining this information requires perturbation almost by definition and in essence represents what has been described as systems biology. It is commonly known as expression proteomics (Blackstock & Weir, 1999) and forms the heart of signal transduction studies, a field that has thrived independently and has been one of the major areas of molecular and cellular biology research for the past 10 years or more (Bradshaw & Dennis, 2003). However, expression proteomics encompasses more than just signal transduction events, even if these are not readily separated from them. These include all nature of metabolic pathways, cell cycling events and intracellular transport, to name only a few. For the most part, efforts in expression proteomics have, to date, been limited to snapshots of the proteome, obtained following an appropriate cellular stimulus, and have concentrated on two kinds of measurements: changes in the level of proteins (up or down) and changes in post-ribosomal modifications, which are usually but not exclusively protein phosphorylations. The former are closely linked to studies using mRNA arrays although formerly these are really a part of transcriptomics.
The final part of the definition can, to complete the analogy, be considered the why of proteomics. This part also encompasses the bioinformatics component of proteomics, another vaguely defined discipline that arose from the realization, as genomics moved into full swing, that the amount of information being obtained was increasing, and would continue to do so in an exponential manner and that managing this inundation was a serious challenge in its own right. Proteomics, far more than genomics and transcriptomics, will accelerate this trend and hence data management must be an integral part of the definition. However, bioinformatics does cut across all of the omics fields, or perhaps more appropriately, provides the fabric that links all of them together so it is not an exclusive part of proteomics either. This last part of the definition really also represents the ultimate goal of proteomics, if a field of study can be said to have a goal, which is to understand not only the nature of the components and how they change with time and condition but also how they integrate to produce a living entity.
Some may argue that this is not an achievable goal either but that is a debate for another time and place.
PROTEOMICS TODAY
Applications - An Overview
Proteomic research legitimately encompasses both classical protein chemistry and the new more complex approaches that feature analyses designed to measure large numbers of proteins at the same time (Aebersold & Mann, 2003, Phizicky et al., 2003), often in a high throughput fashion. Indeed, to many people the latter approaches, which feature such methodology as 2-dimensional polyacrylamide gel electrophoresis (2D PAGE or 2DE), various configurations of mass spectrometry (MS) (most notably applications of electrospray and matrix-assisted laser desorption ionizations [ESI and MALDI]) and the use of arrays (both protein and nucleic acid-based), are proteomics. This uncharitable view diminishes a large amount of important data that was collected primarily over the last half of the previous century and more importantly ignores the fact that the methods and techniques that grew out of that period are still making important contributions to achieving the definitions of Table 1. While new methods often supplant old ones as the one of choice, they rarely if ever eliminate them. Rather they simply expand the arsenal of tools available to answer the experimental question at hand. In this regard it is instructive to briefly compare the foci of the older protein analyses with the newer ones (Table 2). Clearly, the emphasis of earlier studies was on individual protein entities by first striving to obtain homogeneous preparations, initially by isolation of natural proteins and more recently recombinant ones, and then submitting them to detailed characterization including, but certainly not limited to, sequence and structure. These studies were closely coupled to functional analyses that examined kinetic, mechanistic or recognitive properties. This was the core of the reductionist research tradition that produced the basis on which most of modern molecular and cellular biology rests today. In the last few years, the emphasis has shifted in large part to understanding the sociology of proteins, i.e. how these individual entities form a cohesive, responsive (dynamic) network. To use common scientific jargon, this represents a paradigm shift, driven in part by the advances in genomics and transcriptomics and in part by the maturation of 2DE, MS and array technologies. However, it is still of great importance that we understand individual proteins in molecular detail and such studies should not be considered to be outside the scope of proteomics but simply important additional parts of it.
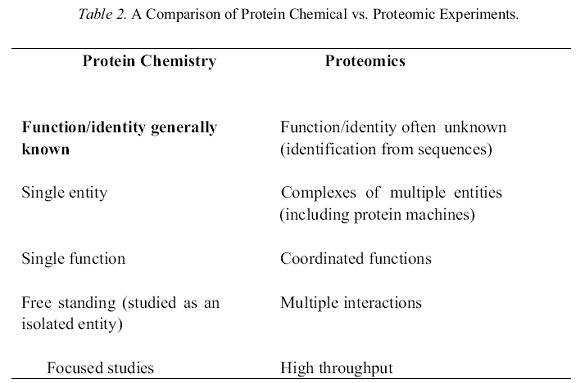
Given this perspective that proteomics is not so much a new field as the substantial expansion of an old one, it is germane to briefly consider where this synthesis of old and new technologies has brought us in terms of some of the more active areas of research before discussing some of the future prospects. Table 3 lists four such areas, which not surprisingly correlate quite closely to the definitions described in Table 1. There is necessarily some overlap between these. For example, both the determination of co- /posttranslational modifications and the identification of protein-protein interactions are also major components of expression profiling in addition to the many modifications and interactions that are not (as yet) directly linked to changes in cell response. Two of these topics are particularly associated with proteomics (protein identification and protein-protein interactions) perhaps because they have provided (or promise to) more new insights than the other areas for which there was already substantial knowledge from independent studies. The ensuing two sections provide brief summaries of some of the advances to date.
1.1 3.2 Protein Identification and Database Mining
From the point of view of one interested in proteomics, there was nothing quite as interesting in the solution of the human (or for that matter any other) genome as the determination that a substantial amount of the putative revealed genes were unknown in both function and three dimensional structure. Although the latter remains true, a number of associative analyses have at least provided a substantial number of indications as to what the general function may be, e.g. DNA repair, even if the exact activity remains to be determined. These have ranged from quite inventive in silico analyses (Eisenberg et al., 2000) that variously take advantage of evolutionary relationships, as revealed by genomic comparisons, to networks established through
protein-protein interactions (see Section 3.3). There nonetheless remains quite large numbers of proteins that still must be identified and characterized. Prof. Sydney Brenner, in a plenary lecture entitled From Genes to Organisms at the Keystone 2000 meeting in Keystone, CO that showcased the leaders in molecular and cellular biology at the turn of the century, noted that if the human genome is composed of 50,000 genes (the number of human genes at that juncture was still quite undecided), it would simply require 50,000 biochemists, each working on one protein product, to define the human proteome. Since he has also been quoted as saying that proteomics is not about new knowledge but about amassing data and that it (proteomics) will prove to be irrelevant
(Cohen, 2001), it isnt clear that he is actually in favor of this. Nonetheless, even though this clearly would not provide all the information required to satisfy the definition given in Table 1, it is not entirely an exaggeration either. We will not make serious headway into human (or other species) proteomics until these unknown gene products are defined and it is hard to see how this wouldnt provide new knowledge.
Aside from determining the nature of the players in the human proteome drama, there is also the considerably larger task of identifying them in real samples. The first major effort to do this was by 2 DE, which was introduced by OFarrell (O'Farrell, 1975) and Klose (Klose, 1975) in the mid 1970s, and arguably marked the beginning of proteomic research. This technique has continued to improve both in reproducibility and sensitivity and has provided a great deal of useful information during the nearly 30 years that it has been available. However, its value is pretty much directly proportional to the ability of the investigator to identify the spots. This has been done in a number of ways but elution and coupling to mass spectrometry has clearly been the most powerful and there are now available well-annotated 2D maps for a number of species and conditions. However, despite the improvements, 2D gels have built in limitations in terms of resolving power and dynamic range that will always limit their usefulness particularly as the focus moves to the lower end of the concentration scale. Thus various chromatographic approaches, such as MudPIT (Wolters et al., 2001) have been introduced to broaden the size and complexity of sample that can be effectively analyzed. These liquid chromatographic systems can be directly linked to the mass spectrometry and in theory can greatly increase the amount of data that can be generated and analyzed. However, only about a third (~1800) of the proteins in a yeast extract have been identified by this approach to date.
There are two basic applications of mass spectrometry used for the identification of proteins; both are dependent on the analysis of generated peptides, usually produced by trypsin (Yates, 1998). In the first, MALDI time-of-flight (TOF) spectra are generated for a mixture of peptides from a pure protein (or relatively simple mixture) and a table of masses is generated that is then matched against similar tables produced from theoretical digests of all proteins listed in the database. For relatively pure samples or simple mixtures, such as would typically be the case with a spot excised from a 2D gel, and where the sample is derived from an organism whose genome is known, this peptide mapping methodology is quite an effective method. However, in cases where the protein sample is derived from a species where the genome may be at best only partially known, its effectiveness falls off rapidly. Of course, co-/posttranslational modifications or unexpected enzyme cleavages will also complicate the picture. Any peptide that contains such alterations or arise from spurious proteolysis will fall outside the theoretical values of the peptides that are computer generated from the database and the coverage (percentage of the complete sequence accounted for in the experimental data set) of the protein will drop. Indeed, one must recover a sizeable portion of the peptides in order that the fit will be statistically significant and this becomes increasingly demanding as databases expand because the number of possible candidates also rises. At the same time, sequence errors in the database will also produce damaging mismatches that will affect the accuracy of such identifications, which unfortunately is a significant problem.
The second method, which generally employs tandem MS (MS/MS) in various configurations, depends on collusion-induced dissociation (CID) events to produce a series of staggered ions that correspond to derivatives of a selected peptide (separated in the initial MS run) that are systematically foreshortened from one end or the other (and then identified in the second MS run). The most useful ions, designated b and y, result from cleavages at the peptide bonds (although the other two types of bonds found in the polypeptide backbone can also be broken as well) and their masses differ successively by the loss of the previous amino acid residue (from one side or the other). Thus a set of b- or y-ions can be directly translated into the sequence of the original peptide. This is clearly far more useful information in making an identification since in theory a sequence of ~10 amino acids should allow a positive identification, particularly if there is added knowledge such as the species of origin. However, in practice, one needs at least two or more sequences from the same protein to be certain of the identification because of a variety of complicating factors (related to converting real data, which is often incomplete, into a theoretical match). This technology is rapidly becoming the method of choice for protein identifications, both for the level of information that it provides and the fact that it is, as noted above, highly adaptable to on-line sampling through direct coupling of liquid chromatography (LC) to the MS/MS instrument.
Both of these methods require facile interaction with and interrogation of protein databases. In the first place, the conversion of the MS/MS data, called de novo sequence interpretation, realistically requires computerized algorithms and search programs since the amount of data that a typical LC MS/MS experiment can generate can run to thousands of spectra. One can interpret MS/MS spectra by hand and then run a BLAST search to make the identification but this is a reasonable approach only if a few spectra are involved, as might be the case for identifying a spot on a 2D gel. For dealing with the large-scale experiments, there are several programs, some of which are publicly available and some of which are not. Some were developed for different MS applications but essentially all can now interpret MS/MS data. There is also related programs available to search for specific features in MS/MS data, such as tyrosine phosphorylation, that are useful, for example, in expression proteomics (Liebler, 2002).
Protein identification permeates all of proteomics and will continue to do so for some time to come. The technology continues to improve and the magnitude and concentration range of samples that can be effectively analyzed has constantly increased for the past decade. There is, however, a long way to go. Consider for example the plasma proteome, long considered a potentially valuable and quite underutilized source of diagnostic information for human disease (Anderson & Anderson, 2002). Although not without its own dynamic aspects and evolving composition, its content is certainly more static than, for instance, the corresponding lysate of a cell. As such, it is clearly a more tractable target. At the same time it is derived from an all-pervasive fluid and as such likely contains products, remnants and detritus indicative of normal and abnormal situations and conditions. The bad news is that that creates an enormous concentration range, and probably number, of analytes. Thus, while the ability to completely define the plasma protein and to facilely assay it would potentially allow physicians to diagnose a far broader spectrum of pathologies than is currently possible, such tests remain a presently unrealized dream. This challenge, among many others like it, is a substantial impetus to continue to develop protein identification methodologies (Petricoin et al.
Protein-protein Interactions
Although the presence of such a substantial number of undefined proteins (in terms of both structure and function) in both simple and complex organismal proteomes was not entirely unexpected, it certainly stimulated (and still does) the imagination and enthusiasm of life scientists. If anything, the revelation of the extent to which intracellular protein-protein interactions occur has done so to an even greater extent. It is perhaps the greatest surprise that proteomics has produced to date.
Protein chemists have long been aware that most intracellular enzymes are oligomeric (dimers and tetramers being strongly favored) but there was little evidence from earlier studies for extensive interactions beyond that. However, to be fair, the types of experiments and the questions being asked then did not lend themselves, for the most part, to detecting them. With the availability of recombinant proteins in the 1980s, this scenario started to change and in the early 1990s, for example, signal transduction experiments began to show the formation of substantial complexes induced by and associated with activated receptors. Similar observations were made regarding the transcriptional machinery as well as many other cellular complexes. These experiments often utilized antibodies to precipitate a putative member of a complex and were then analyzed, after separation on gels, by additional immuno-reagents in Western blot format. Increasingly MS was also used to identify the partners as new entities, for which antibodies were not available at the time, were discovered. These pulldowns were quite instructive and a goodly number of the known signal transducers (scaffolds, adaptors and effectors), for example, were so identified. Immunological recognition still remains a powerful tool in many proteomic applications. However, it was not until more non-specific methodology was employed that the vast scope and range of intracellular protein interactions became evident. This began with yeast 2-hybrid analyses
(Fields & Song, 1989) and then branched out into MS-tag methods (where a member of the complex is labeled or tagged with an entity that can be detected by an antibody or other affinity reagent) (Aebersold & Mann, 2003); both were soon adapted to high-throughput (HTP) capability that substantially ratcheted up the number of interactions identified. The results have been impressive although in no case are they complete.
Although
these kinds of studies have been applied to a number of research paradigms, the
most extensive analyses have been made with yeast. The resultant development of
the yeast protein interaction map is well illustrated by four seminal articles
(Gavin et al.,
2002, Ho et al.,
2002, Ito et al.,
2001, Uetz et al.,
2000) (and a fifth that summarizes the collected findings of these and related
studies (von Mering et al.,
2002)). The first two (Ito et al.,
2001, Uetz et al.,
2000) described the application of scaled up 2-hybrid analyses, accounting
collectively for over 5000 binary interactions, while the latter two used two
forms of MS-tag technology that, when reduced to binary interactions, accounted
for about 18,000 and 33,000 assignments (von Mering et al., 2002). The
first of these (Gavin et al.,
2002) used tandem-affinity purification (TAP); the second (Ho et al., 2002) was based
on the expression of a large number of tagged bait proteins expressed in
yeast and was called high-throughput mass spectrometric protein complex
identification (HMS-PCI). It is important to note that the latter two methods
are meant to trap and identify larger complexes containing multiple partners
while the 2-hybrid method is designed to identify only interacting pairs. In
comparing these results, von Mering et al. (von Mering et al., 2002) also
included interactions detected indirectly, i.e. from gene expression (chip)
and genetic lethality data, and predicted from various genomic analyses
(Eisenberg et al.,
2000). They were all compared against a reference standard of some 10,900
interactions culled from manually curated catalogues that were derived from
the
These values and conclusions are both exciting and disappointing. On the one hand, they suggest that the copious number of interactions already observed is representative of a detailed intracellular protein network through which metabolic and informational fluxes flow to support the essential life processes. However, on the other hand, they clearly indicate the inadequacies of the present methodologies. Although these HTP methods have produced masses of data, large parts of it are apparently spurious or at least questionable and, perhaps even more troubling, they are not detecting even the majority of presumably relevant interactions. Clearly there is a long way to go in this area as well.
ARRAYS, CHIPS AND HIGH-THROUGHPUT - IS
BIGGER BETTER ?
Regardless of the nature of the proteome, proteomic analyses are inherently large in scope (Table 2) but at the same time offer the opportunity to look at biology (or biological systems) on a different and more complex level (Phizicky et al., 2003). However, size also offers challenges and these have been met, at least initially, by the introduction of HTP methods (or ones that hold the promise to be adaptable to HTP). Chief among these methods are arrays, ensembles of molecules of different types that can be built with robots and interrogated with large-scale query sets of various types. Indeed, some consider that arrays are proteomics (Petach & Gold, 2002) and
apparently leave every thing else for (modern?) protein chemistry.
Intellectually, protein arrays are a direct extension of nucleic acid arrays (or chips) that display cDNAs (or corresponding oligonucleotides) derived from a germane library that can be probed with mRNA prepared from cells that have been stimulated in some fashion. This has become the premier way to examine gene expression (Shalon et al., 1996) and has been useful in proteomics to compare transcript and protein levels and to identify proteinprotein interactions among other things. In the main its value is to reveal patterns that can be related to the causative stimulus or underlying pathology of the test cell and it has shown some diagnostic potential. Protein arrays are, however, more complex. They can be built by immobilizing either the test molecules, sometimes designated analyte specific reagents (ASR) (Petach & Gold, 2002), or the proteins to be tested. The means of attachment also vary tremendously reflecting the greater difficulty in attaching native proteins and maintaining their three-dimensional structure throughout the analyses. ASRs include such moieties as antibodies (Hanash, 2003), peptide and nucleic acid aptamers (Geyer & Brent, 2000, Petach & Gold, 2002), and protein domains (Nollau & Mayer, 2001), e.g., the SH2 domain that recognizes phosphotyrosine-containing sequences in signal transduction systems. Protein arrays reflecting a (nearly) whole proteome have also been prepared and these can be used to identify function, protein-protein interactions, etc. (Grayhack & Phizicky, 2001, Zhu et al. New and
inventive protein arrays appear with regularity and their use will be a major part of tomorrows proteomics.
As exciting as these approaches are, and the volume of new information they produce notwithstanding, they are neither comprehensive nor error free. Indeed, as with the TAP and HMS-PCI data (see Section 3.3), it is not until the data have been substantially filtered and reconfirmed (by independent means) do they begin to become really reliable. That seems to be something of a norm for all HTP experiments. Thus, one might reasonably ask if trying to examine too bigger picture may not be at least in part counter productive. The response to this really depends on the objective of the experiment. If large-scale screening to find new drug targets is the purpose, any identification will ultimately be checked and cross-checked during the target validation phase. However, how are the vast numbers of protein-protein interactions in say a HMS-PCI experiment to be checked? Ultimately each will have to be verified (and undoubtedly will be) but almost certainly by a process that is slower, more exact but more time consuming. Large-scale array experiments have and will continue to provide ever broader views into biology but their contribution to the fine details will, at least for the moment, likely be considerable less.
WITHER PROTEOMICS ?
Few fields have emerged with as much promise as proteomics, and with the possible exception of genomics, as much hype (Cohen, 2001). Even casual assessments have suggested that neither biology nor medicine will ever be quite the same again (as proteomics takes root) and that is pretty strong stuff. However, it is not necessarily wrong or even exaggerated. The problem of course is not in the field, for it is easy to realize that if the proteomic definitions given in Table 1 are even modestly achieved the effects on animal and human health care, food production and world ecology will be enormous, but rather in our ability to make the needed measurements. The problems are partly technological and partly intellectual. Chief among the technical problems is the dynamic (concentration) range of samples, quantification (or lack thereof), and information (overload) management. However, it may be expected that in time these and other technological hurdles will be over come; one cannot be quite so sure about the intellectual barriers.
Some insight into this latter issue comes from a comparison of the manned space program of NASA, which first placed a human on the moon in 1969, and the determination of the human genome, which was completed in the last year (or so). When the decision to reach both of these goals was taken, the only significant obstacles were technological and in both cases highly inventive minds produced the needed breakthroughs. As a result, both stand as monumental human achievements. However, the promises of the manned space program have not continued to live up to the initial event in terms of accomplishment or return of new knowledge. That is not to say that space studies have not yielded nor will continue to yield important advances; it is only the gap between expectation and reality that is disappointing. Perhaps colonies on the moon or manned exploration of Mars were not really realistic so quickly but such thoughts were certainly entertained and, more importantly, expressed after the first lunar landing. Since in this analogy functional genomics represents the lunar colonies, i.e. the longterm promise, and since proteomics is substantially synonymous with functional genomics, it follows that the burden of capitalizing on the human genome project falls largely to proteomics. And this is where the intellectual side appears. NASA has suffered enormously from all manner of over-analyses, priority changes and shifting political winds and agendas, in addition to continued daunting technical challenges, and these quite human, but largely non-scientific activities have sapped its resources and opportunities. The same could certainly happen to proteomics. A few concerns are illustrative.
Much of proteomics, and indeed much of its lure, lies in the fact that so much of the proteome - any proteome - is unknown. However, mapping unknown functions and elucidating new protein-protein interactions is not hypothesis driven research and funding agencies, particularly in the US, have pretty much eschewed anything that is not. Perhaps because discovery research has been largely the province of industry, particularly the pharmaceutical industry, grant reviewers deem it somehow an inappropriate approach. Just how these same people think that these data, which are crucial for all proteomic research, and arguably all biological research, are going to be obtained remains a mystery although it seems likely that at least some of the information will be derived from industrial research (in part because academic laboratories will be shut out of this activity, particularly smaller ones) (Cohen, 2001). This will contribute to a related problem, to wit the control of intellectual property and the need to place proteomic data in the public domain. There are no easy solutions to this complex problem and it promises to be a significant deterrent. An internationally funded public human proteome project would be a very worthy investment for the worlds governments. It would have to have more tightly defined objectives than the global definitions (Table 1) given above but just coordinating the various activities underway would be a good first step (Tyers & Mann, 2003).
Nowhere is the promise of proteomics greater than in clinical applications (Hanash, 2003). The ensuing chapters of this volume discuss these at length, illustrating the degree to which proteomics is already contributing to medical care and providing signposts for the future directions. As basic research, drug discovery and diagnostic and therapeutic applications coalesce, the true potential of proteomics will become clear. Let us hope that it is of the magnitude and significance predicted in the following pages.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 1208
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved