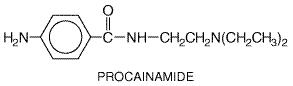| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Antiarrhythmic Drugs
Overview
|
Individual cardiac cells undergo depolarization and repolarization to form cardiac action potentials about sixty times per minute. The shape and duration of each action potential are determined by the activity of ion channel protein complexes on the surface of individual cells, and the genes encoding many of these proteins have now been identified. Thus, each heartbeat is the result of the highly integrated electrophysiological behavior of multiple gene products on multiple cardiac cells. Ion channel function can be perturbed by factors such as acute ischemia, sympathetic stimulation, or myocardial scarring to create abnormalities of cardiac rhythm, or arrhythmias. Available antiarrhythmic drugs suppress arrhythmias by blocking flow through specific ion channels or by altering autonomic function. Arrhythmias can range from incidental, asymptomatic clinical findings to life-threatening abnormalities. Mechanisms underlying cardiac arrhythmias have been identified in cellular and animal experiments. In some human cases, precise mechanisms are known, and treatment targeted against those mechanisms can be used. In other cases, mechanisms can be only inferred, and the choice of drugs is based largely on results of prior experience. Antiarrhythmic drug therapy can have two goals: termination of an ongoing arrhythmia or prevention of an arrhythmia. It is now well recognized that antiarrhythmic drugs not only help to control arrhythmias but also can cause them, especially during long-term therapy. Thus, prescribing antiarrhythmic drugs requires that precipitating factors be excluded or minimized, that a precise diagnosis of the type of arrhythmia (and its possible mechanisms) be made, that the prescriber has reason to believe that drug therapy will be beneficial, and that the risks of drug therapy be minimized. In this chapter, the principles underlying normal and abnormal cardiac electrophysiology are outlined. Then, the mechanisms by which drugs modulate cardiac electrophysiology are presented, followed by a description of the important properties of individual agents. |
Principles of Cardiac Electrophysiology
|
The flow of charged ions across cell membranes results in the ionic currents that make up cardiac action potentials. The factors that determine the magnitude of individual currents and how these are modified by drugs now can be elucidated at the cellular and molecular levels (Fozzard and Arnsdorf, 1991; Snyders et al., 1991; Priori et al., 1999). However, the action potential is a highly integrated entity: changes in one current almost inevitably produce secondary changes in other currents. Most antiarrhythmic drugs affect more than one ion current, and many exert ancillary effects such as modification of cardiac contractility or autonomic nervous system function. Thus, antiarrhythmic drugs usually exert multiple actions and can be beneficial or harmful in individual patients (Roden, 1994; Priori et al., 1999). The Cardiac Cell at Rest: A K+-Permeable Membrane Ions move across cell membranes in response to electrical and concentration gradients, not through the lipid bilayer but through specific ion channels or transporters. The normal cardiac cell at rest maintains a transmembrane potential approximately 80 to 90 mV negative to the exterior; this gradient is established by pumps, especially Na+,K+ATPase, and fixed anionic charges within cells. There is both an electrical and a concentration gradient that would move Na+ ions into resting cells (Figure 351). However, Na+ channels, which allow Na+ to move along this gradient, are closed at negative transmembrane potentials, so Na+ does not enter normal resting cardiac cells. In contrast, a specific type of K+ channel protein (the inward rectifier channel) is in an open conformation at negative potentials: i.e., K+ can move across the cell membrane at negative potentials in response to either electrical or concentration gradients (Figure 351). For each individual ion, there is an equilibrium potential Ex at which there is no net driving force for the ion to move across the membrane. Ex can be calculated using the Nernst equation:
Ex=61 log ([x]i/[x]o) (351) where [x]o is the extracellular concentration of the ion and [x]i is the intracellular concentration. For usual values for K+, [K]o= 4 mM and [K]i= 140 mM, the calculated K+ equilibrium potential EK is 94 mV. There is thus no net force driving K+ ions into or out of a cell when the transmembrane potential is 94 mV, which is close to the resting potential. If [K]o is elevated to 10 mM, as might occur in diseases such as renal failure or myocardial ischemia, the calculated EK rises to 70 mV. In this situation, K+ will tend to move down its concentration gradient. In fact, there is excellent agreement between changes in theoretical EK due to changes in [K]o and the actual measured transmembrane potential (Figure 352), indicating that the normal cardiac cell at rest is permeable to K+ (because inward rectifier channels are open) and that the concentration of K+ in the extracellular space is the major determinant of resting potential.
Na+ Channel Opening Initiates the Action Potential: Ion Currents If a cardiac cell at rest is depolarized above a threshold potential, Na+ channel proteins change conformation from the 'closed' (or rest) to the conducting ('open') state, allowing up to 107 Na+ ions per second to enter each cell and moving the transmembrane potential towards ENa (+65 mV). This surge of Na+ ion movement lasts only about a millisecond, after which the Na+ channel protein rapidly changes conformation from the 'open' state to an 'inactivated,' nonconducting state. Measuring Na+ current directly is technically demanding; therefore many studies report the maximum upstroke slope of phase 0 (dV/dtmax, or Vmax) of the action potential (Figure 353), which is proportional to Na+ current. The traditional view is that Na+ channels, once inactivated, cannot reopen until they reassume the rested, or closed, conformation. Electrophysiological techniques capable of measuring the behavior of individual ion channel proteins are now revealing some of the detailed mechanisms of these state transitions, and the findings obtained are changing some traditional views. For example, a small population of Na+ channels may continue to open during the action potential plateau in some cells (Figure 353). In fact, a defect in the structural region of the Na+ channel protein that has been implicated in control of channel inactivation is responsible for one form of the congenital long QT syndrome, a disease associated with abnormal repolarization and serious arrhythmias (Roden and Spooner, 1999). In general, however, as the cell membrane repolarizes, the changes in membrane potential to which Na+ channel proteins are subject moves them from inactivated to 'closed' conformations. The relationship between Na+ channel availability and transmembrane potential is an important determinant of conduction and of refractoriness in many cells, as discussed below.
The changes in transmembrane potential generated by the inward Na+ current produce, in turn, a series of openings (and in some cases subsequent inactivation) of other channels (Figure 353). For example, when a cell from the epicardium or the HisPurkinje conducting system is depolarized by the Na+ current, 'transient outward' K+ channels change conformation to enter an open, or conducting, state; since the transmembrane potential at the end of phase 0 is positive to EK, the opening of transient outward channels results in an outward, or repolarizing, K+ current (termed ITO), which contributes to the phase 1 'notch' seen in some action potentials. Transient outward K+ channels, like Na+ channels, rapidly inactivate. During the phase 2 plateau of a normal cardiac action potential, an inward, depolarizing current primarily through Ca2+ channels is balanced by an outward, repolarizing current primarily through K+ ('delayed rectifier') channels. Delayed rectifier currents (termed IK) increase with time, while Ca2+ currents inactivate (and so decrease with time); the result is repolarization of the cardiac cell (phase 3) several hundred milliseconds after the initial Na+ channel opening. Mutations in the genes encoding repolarizing K+ channels are responsible for other forms of the congenital long QT syndrome (Roden and Spooner, 1999). Identification of these specific channels has allowed more precise characterization of the pharmacological effects of antiarrhythmic drugs. A common mechanism whereby drugs prolong cardiac action potentials is inhibition of specific delayed rectifier current, IKr. Differing Action Potential Behaviors Among Cardiac Cells This general description of the action potential and the currents that underlie it must be modified for certain cell types (Figure 354), presumably because of variability in the number or products of ion channel genes expressed in individual cells. Endocardial ventricular cells lack a prominent transient outward current, while cells from the subendocardial HisPurkinje conducting system (and in some species from the midmyocardium) have very long action potentials (Antzelevitch et al., 1991). Atrial cells have very short action potentials, probably because ITO is larger, and an additional repolarizing K+ current, activated by the neurotransmitter acetylcholine, is present. As a result, vagal stimulation further shortens atrial action potentials. Cells of the sinus and atrioventricular (AV) nodes lack substantial Na+ currents. In addition, these cells, as well as cells from the conducting system, normally display the phenomenon of spontaneous diastolic, or phase 4, depolarization and thus spontaneously reach threshold for regeneration of action potentials. The rate of spontaneous firing usually is fastest in sinus node cells, which therefore serve as the natural pacemaker of the heart. Specialized K+ channels underlie the pacemaker current in heart.
Modern molecular biological and electrophysiological techniques, by which the behavior of single ion channel proteins in an isolated patch of membrane can be studied, have refined the description of ion channels important for the normal functioning of cardiac cells and have identified channels that may be particularly important under pathological conditions. For example, it is now established that transient outward and delayed rectifier currents actually result from multiple ion channel subtypes (Figure 353; Tseng and Hoffman, 1989; Sanguinetti and Jurkiewicz, 1990), and that acetylcholine-evoked hyperpolarization results from activation of a K+ channel formed by hetero-oligomerization of multiple, distinct channel proteins (Krapivinsky et al., 1995). The understanding that molecularly diverse entities subserve regulation of the cardiac action potential is important because drugs may target one channel subtype selectively. Furthermore, ancillary function-modifying proteins (the products of diverse genes) have been identified for most ion channels. In addition to the usual ('L-type') Ca2+ channels, a second type of Ca2+ channel, which is most prominent at relatively negative potentials, has been identified in some cardiac cells (Bean, 1985). This 'T-type' Ca2+ channel may be important in diseases such as hypertension and may play a role in pacemaker activity in some cells. A T-typeselective antihypertensive agent, mibefradil, was available briefly in the late 1990s but was withdrawn because it was involved in many serious, undesirable drugdrug interactions. Specific channels that transport Cl ions and result in repolarizing currents (ICl) have been identified in many species (Hume and Harvey, 1991); some of these are observed only under pathophysiological conditions, such as adrenergic stimulation. Some K+ channels are quiescent when intracellular ATP stores are normal and become active when these stores are depleted. Such ATP-inhibited K+ channels may become particularly important in repolarizing cells during states of metabolic stress such as myocardial ischemia (Weiss et al., 1991; Wilde and Janse, 1994). Maintenance of Intracellular Homeostasis With each action potential, the cell interior gains Na+ ions and loses K+ ions. An ATP-requiring Na+K+ exchange mechanism, or pump, is activated in most cells to maintain intracellular homeostasis. This Na+,K+ATPase extrudes three Na+ ions for every two K+ ions shuttled from the exterior of the cell to the interior; as a result, the act of pumping itself generates a net outward (repolarizing) current. Normally, intracellular Ca2+ is maintained at very low levels (<100 nM). In heart cells, the entry of Ca2+ during each action potential is a signal to the sarcoplasmic reticulum to release its Ca2+ stores. The resultant increase in intracellular Ca2+ then allows Ca2+-dependent contractile processes to occur. Removal of intracellular Ca2+ occurs by both an ATP-dependent Ca2+ pump (which moves Ca2+ ions back to storage sites in the sarcoplasmic reticulum) and an electrogenic Na+Ca2+ exchange mechanism on the cell surface, which exchanges three Na+ ions from the exterior for each Ca2+ ion extruded. The initial rise in Ca2+, which serves as the trigger for Ca2+ release from intracellular stores, is a result of the opening of Ca2+ channels in the cell membrane or of Ca2+ entry through Na+Ca2+ exchange; i.e., in response to phase 0 entry of Na+, the Na+Ca2+ exchange protein may transiently extrude Na+ ions in exchange for Ca2+ ions (Figure 353). Impulse Propagation and the Electrocardiogram Normal cardiac impulses originate in the sinus node. Impulse propagation in the heart depends on two factors: the magnitude of the depolarizing current (usually Na+ current) and the geometry of cellcell electrical connections. Cardiac cells are long and thin and well coupled through specialized gap junction proteins at their ends, whereas lateral ('transverse') gap junctions are sparser. As a result, impulses spread along cells two to three times faster than across cells. This 'anisotropic' (direction-dependent) conduction may be a factor in the genesis of certain arrhythmias described below (Priori et al., 1999). Once impulses leave the sinus node, they propagate rapidly throughout the atria, resulting in atrial systole and the P wave of the surface electrocardiogram (ECG; Figure 354). Propagation slows markedly through the AV node, where the inward current (through Ca2+ channels) is much smaller than the Na+ current in atria, ventricles, or the subendocardial conducting system. This conduction delay allows the atrial contraction to propel blood into the ventricle, thereby optimizing cardiac output. Once impulses exit from the AV node, they enter the conducting system, where Na+ currents are larger than in any other tissue. Hence, propagation is correspondingly faster, up to 0.75 meter/second longitudinally, allowing coordinated ventricular contraction, the QRS complex on the ECG, as impulses spread from the endocardium to the epicardium. Ventricular repolarization results in the T wave of the ECG. The ECG can be used as a rough guide to some cellular properties of cardiac tissue (Figure 354): (1) heart rate reflects sinus node automaticity, (2) PR interval duration reflects AV nodal conduction time, (3) QRS duration reflects conduction time in the ventricle, and (4) the QT interval is a measure of ventricular action potential duration. Refractoriness: Fast-Response Versus Slow-Response Tissue If a single action potential, such as that shown in Figure 353, is restimulated very early during the plateau, no Na+ channels are available to open, so no inward current results and no action potential is generated: the cell is refractory. If, on the other hand, an extrastimulus occurs after the cell has repolarized completely, Na+ channels have recovered from inactivation, and a normal Na+ channeldependent upstroke results (Figure 355A). When an extrastimulus occurs during phase 3 of the action potential, the magnitude of the resultant Na+ current is dependent on the number of Na+ channels that have recovered from inactivation (Figure 355A), which, in turn, is dependent on the voltage at which the extrastimulus was applied. Thus, in atrial, ventricular, and HisPurkinje cells ('fast-response cells'), refractoriness is determined by the voltage-dependent recovery of Na+ channels from inactivation. Refractoriness also frequently is measured by assessing whether premature stimuli applied to tissue preparations (or the whole heart) result in propagated impulses. While the magnitude of the Na+ current is one major determinant of such propagation, cellular geometry (see above) also is important in multicellular preparations. Ordinarily, each cell is connected to many neighbors, so that impulses spread rapidly, and the heart acts like a single large cell, a 'syncytium.' However, if the geometric arrangement is such that a single cell must supply depolarizing current to many neighbors, conduction can fail. The effective refractory period (ERP) is the shortest interval at which a premature stimulus results in a propagated response and is often used to describe drug effects in intact tissue.
The situation is different in Ca2+ channeldependent ('slow-response') tissue such as the AV node. The major factor controlling recovery from inactivation of Ca2+ channels is time (Figure 355C). Thus, even after a Ca2+ channeldependent action potential has repolarized back to its initial resting potential, Ca2+ channels are not all available for reexcitation. Therefore, an extrastimulus applied shortly after repolarization is complete results in a reduced Ca2+ current, which may propagate slowly to adjacent cells prior to extinction. An extrastimulus applied later will result in a larger Ca2+ current and faster propagation. Thus, in Ca2+ channeldependent tissues, which include not only the AV node but also tissues whose underlying characteristics have been altered by factors such as myocardial ischemia, refractoriness is time-dependent, and propagation occurs slowly. Conduction that exhibits such dependence on the timing of premature stimuli is termed 'decremental.' By contrast, conduction velocity is independent of prematurity in fast-response tissues until a stimulus shorter than the effective refractory period is applied, when it fails completely ('all-or-none' response). Slow conduction in the heart, a critical factor in the genesis of reentrant arrhythmias (below), also can occur when Na+ currents are depressed by disease or membrane depolarization (e.g., elevated [K]o), resulting in decreased steady-state Na+ channel availability (Figure 355B). |
Mechanisms of Cardiac Arrhythmias
|
When the normal sequence of impulse initiation and propagation is perturbed, an arrhythmia occurs. Failure of impulse initiation may result in slow heart rates (bradyarrhythmias), and failure of impulses to propagate normally from atrium to ventricle results in dropped beats or 'heart block,' which usually reflects an abnormality in either the AV node or the HisPurkinje system. These abnormalities may be caused by drugs (Table 351) or by structural heart disease; in the latter case, permanent cardiac pacing may be required. Abnormally rapid heart rhythms (tachyarrhythmias) are common clinical problems that may be treated with antiarrhythmic drugs. Three major underlying mechanisms have been identified: enhanced automaticity, triggered automaticity, and reentry. Enhanced Automaticity Enhanced automaticity may occur in cells that normally display
spontaneous diastolic depolarizationthe sinus and AV nodes and the
HisPurkinje system. Afterdepolarizations and Triggered Automaticity Under some pathophysiological conditions, a normal cardiac action
potential may be interrupted or followed by an abnormal depolarization (Figure
356). If this abnormal depolarization reaches threshold, it may, in turn,
give rise to secondary upstrokes which then can propagate and create abnormal
rhythms. These abnormal secondary upstrokes occur only after an initial normal,
or 'triggering,' upstroke and so are termed triggered rhythms.
Two major forms of triggered rhythms are recognized: (1) Under conditions of
intracellular Ca2+ overload (myocardial ischemia, adrenergic
stress, digitalis intoxication), a normal action potential may be followed by
a 'delayed afterdepolarization' (DAD; Figure 356A). If this
afterdepolarization reaches threshold, a secondary triggered beat or beats
may occur. DAD amplitude is increased in vitro by rapid pacing, and
clinical arrhythmias thought to correspond to DAD-mediated triggered beats
are more frequent when the underlying cardiac rate is rapid (Rosen and Reder,
1981). (2) The key abnormality in the second type of triggered activity is
marked prolongation of the cardiac action potential. When this occurs, phase
3 repolarization may be interrupted by an 'early
afterdepolarization' (EAD; Figure 356B). EAD-mediated triggering
in vitro and clinical arrhythmias are most common when the underlying
heart rate is slow, extracellular K+ is low, and certain drugs
(antiarrhythmics and others) that prolong action potential duration are
present. EADs represent, by definition, an increase in net inward current
during repolarization. However, it is not certain through which channel(s)
current flows to generate EADs. When an EAD is present, sympathetic
stimulation (
Reentry Anatomically Defined Reentry The principles of cardiac reentry were first described early in the 1900s. Reentry can occur when impulses propagate by more than one pathway between two points in the heart and those pathways have heterogeneous electrophysiological properties. An 'experiment of nature' is the WolffParkinsonWhite (WPW) syndrome, described in the 1930s. Patients with WPW have accessory connections between the atrium and ventricle (Figure 357). With each sinus node depolarization, impulses can excite the ventricle via the normal structures (AV node) or the accessory pathway. However, the electrophysiological properties of the AV node and accessory pathways are different: accessory pathways consist of fast-response tissue, whereas the AV node is composed of slow-response tissue. Thus, with a premature atrial beat, conduction may fail in the accessory pathway and continue to conduct, albeit slowly, in the AV node and then through the HisPurkinje system, where the propagating impulse may encounter the ventricular end of the accessory pathway when it is no longer refractory. Note that the likelihood that the accessory pathway is no longer refractory increases as AV nodal conduction slows. When the impulse reenters the atrium, it can then reenter the ventricle via the AV node, reenter the atrium via the accessory pathway, and so on (Figure 357). Reentry of this type is therefore determined by (1) the presence of an anatomically defined circuit, (2) heterogeneity in refractoriness among regions in the circuit, and (3) slow conduction in one part of the circuit. Similar 'anatomically defined' reentry commonly occurs in the region of the AV node (AV nodal reentrant tachycardia) and in the atrium (atrial flutter). The term paroxysmal supraventricular tachycardia (PSVT) includes both AV reentry and AV nodal reentry, which share many clinical features. In some of these cases, it is now possible to identify and nonpharmacologically ablate critical portions of reentrant pathways (or automatic foci), thus curing the patient and obviating the need for long-term drug therapy. The procedure is carried out through a catheter advanced to the interior of the heart and requires minimal convalescence.
Functionally Defined Reentry Reentry also may occur in the absence of a distinct, anatomically defined pathway (Figure 358). For example, alterations in cellcell coupling following acute myocardial infarction in dogs result in reentrant ventricular tachycardia (VT) whose circuit is dependent not only on postinfarction scarring but also on the rapid longitudinal and slow transverse conduction properties of cardiac tissue (Wit et al., 1990). If ischemia or other electrophysiological perturbations result in an area of sufficiently slow conduction in the ventricle, impulses exiting from that area may find the rest of the myocardium reexcitable, in which case fibrillation may ensue. Atrial or ventricular fibrillation is an extreme example of 'functionally defined' (or 'leading circle') reentry: cells are reexcited as soon as they are repolarized sufficiently to allow enough Na+ channels to recover from inactivation. In this setting, neither organized activation patterns nor coordinated contractile activity is present.
Common Arrhythmias and Their Mechanisms The primary tool for diagnosis of arrhythmias is the
electrocardiogram, although more sophisticated approaches, such as recording
from specific regions of the heart during artificial induction of arrhythmias
by specialized pacing techniques, sometimes are used. Table 352 lists common
arrhythmias, their likely mechanisms, and approaches that should be
considered for their acute termination and for long-term therapy to prevent
recurrence. Examples of some arrhythmias discussed here are shown in Figure
359. Some arrhythmias, notably ventricular fibrillation (VF), are best
treated not with drugs but with DC cardioversionthe application of a large
electric current across the chest. This technique also can be used to
immediately restore normal rhythm in less serious cases; if the patient is
conscious, a brief period of general anesthesia is required. Implantable
cardioverter/defibrillators (ICDs), devices that are capable of detecting VF
and automatically delivering a defibrillating shock, increasingly are being
used in patients who have been resuscitated from one episode of VF. Often
drugs are used with these devices to reduce the need
|
Mechanisms of Antiarrhythmic Drug Action
|
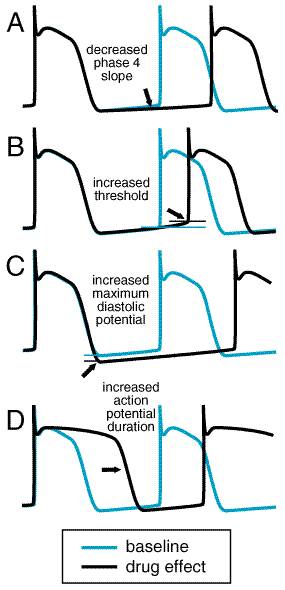
Drug effects that may be antiarrhythmic can be demonstrated in vitro or in animal models, but the relationship between the multiple effects these drugs produce in patients and their effects on arrhythmias (whose mechanisms are only sometimes known) can be complex. A single arrhythmia may result from multiple mechanisms; for example, an automatic or triggered beat may result in a sustained reentrant arrhythmia in a patient with a potential reentrant circuit. Drugs may be antiarrhythmic by suppressing the initiating mechanism or by altering the reentrant circuit. In some cases, however, drugs also may suppress the initiator but nonetheless promote reentry (see below). Drugs may slow automatic rhythms by altering one of the four
determinants of spontaneous pacemaker discharge (Figure 3510): maximum
diastolic potential, phase 4 slope, threshold potential, or action potential
duration. Block of Na+ or Ca2+ channels usually results
in altered threshold, block of cardiac K+ channels prolongs action
potential, adenosine and acetylcholine may increase maximum diastolic
potential, and
Antiarrhythmic
drugs may block arrhythmias due to DADs or EADs through two major mechanisms:
(1) inhibition of the development of afterdepolarizations or (2) interference
with the inward current (usually through Na+ or Ca2+
channels), which is responsible for the upstroke. Thus, for example,
arrhythmias due to digitalis-induced DADs may be inhibited by verapamil
(which blocks the development of DAD) or by quinidine (which blocks Na+
channels, thus elevating the threshold required to produce the abnormal
upstroke). Similarly, two approaches are used in arrhythmias thought to be
related to EAD-induced triggered beats (Tables 351 and 352). EADs can be
inhibited by shortening action potential duration; in practice, heart rate is
accelerated by isoproterenol infusion or by pacing. Triggered beats arising
from EADs can be inhibited by Mg2+, without normalizing
repolarization in vitro or QT interval in patients, through mechanisms
that are not well understood. In patients with a congenitally prolonged QT
interval, torsades de pointes often occur with adrenergic stress;
preventive treatment includes In anatomically determined reentry, drugs may terminate the arrhythmia
by blocking propagation of the action potential. Conduction usually fails in
a 'weak leak' in the circuit. In the example of the WPW-related
arrhythmia described above, the weak link is the AV node, and drugs that
prolong AV nodal refractoriness and slow AV nodal conduction, such as Ca2+
channel blockers,
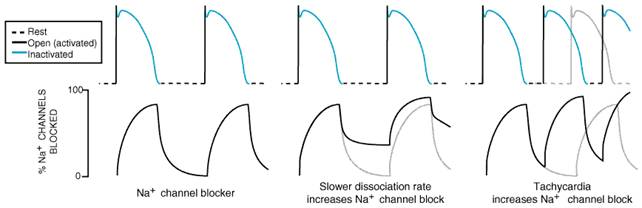
State-Dependent Ion Channel Block A key concept in understanding differences in the clinical actions among antiarrhythmic drugs is state-dependent block. Experimental evidence strongly supports the idea that ion channelblocking drugs bind to specific receptor-like sites on the ion channel proteins to modify function (e.g., decrease current) and that, as an ion channel protein shuttles among functional conformations (or ion channel 'states'), the affinity of the ion channel protein for the drug on its target site will vary (Hille, 1977; Hondeghem and Katzung, 1984; Snyders et al., 1991). Drugs are thought to gain access to target sites by at least two routes: through the pore (the 'hydrophilic' pathway) and through the lipid bilayer (the 'hydrophobic' pathway). Physicochemical characteristics, such as molecular weight or lipid solubility, are important determinants of state-dependent binding. State-dependent binding has been studied most extensively in the case of Na+ channelblocking drugs. Most useful agents of this type block open and/or inactivated Na+ channels and have very little affinity for channels in the resting state. Thus, with each action potential, drugs bind to Na+ channels (and block them), and with each diastolic interval, drugs dissociate and 'unblock.' Block may be due to a drug binding within the conduction pore or to binding at a remote site that then induces changes in the ability of the channel protein to form a pore (an 'allosteric' effect). As illustrated in Figure 3512, the unblocking rate is a key determinant of steady-state block of Na+ channels. When heart rate increases, the time available for unblocking decreases, and steady-state Na+ channel block increases. The rate of recovery from block also slows as cells are depolarized, as in ischemia (Chen et al., 1975). This provides the basis for the finding that Na+ channel blockers depress Na+ current, and hence conduction, to a greater extent in ischemic tissues than in normal tissues. Open-versus inactivated-state block also may be important in determining the effects of some drugs. For example, increased action potential duration, which results in a relative increase in time spent in the inactivated state, may increase block by drugs such as lidocaine or amiodarone, which bind to inactivated channels (Hondeghem and Katzung, 1984). The rate of recovery from block often is expressed as a time constant
( Classifying Antiarrhythmic Drugs Classifying drugs by common electrophysiological properties emphasizes the connection between basic electrophysiological actions and antiarrhythmic effects (Vaughan Williams, 1992). To the extent that the clinical actions of drugs can be predicted from their basic electrophysiological properties, such classification schemes have some merit. However, as each compound is better characterized in a range of in vitro and in vivo test systems, it becomes apparent that, even among drugs that share the same classification, differences in pharmacological effects occur, some of which may be responsible for the observed clinical differences in responses to drugs of the same broad 'class' (Table 353). An alternative way of approaching antiarrhythmic therapy is to attempt to classify arrhythmia mechanisms and then to target drug therapy to the electrophysiological mechanism most likely to terminate or prevent the arrhythmia (Table 352; Task Force, 1991). Na+ Channel Block The extent of Na+ channel block is critically dependent on
heart rate and membrane potential as well as on drug-specific physicochemical
characteristics that determine By increasing threshold, Na+ channel block decreases
automaticity (Figure 3510B) and can inhibit triggered activity
arising from delayed afterdepolarizations (DADs) or early afterdepolarizations
(EADs). Many Na+ channel blockers also decrease phase 4 slope (Figure
3510A). In anatomically defined reentry, Na+ channel
blockers may decrease conduction sufficiently to extinguish the propagating
reentrant wavefront. However, as described above, conduction slowing due to
Na+ channel block may exacerbate reentry. Block of Na+
channels also shifts the voltage dependence of recovery from inactivation (Figure
355B) to more negative potentials, thereby tending to increase
refractoriness. Thus, whether a given drug exacerbates or suppresses a
reentrant arrhythmia depends on the balance between its effects on
refractoriness and on conduction in a particular reentrant circuit. In most
studies, Na+ channel blockers prevent recurrence of reentrant
ventricular tachycardia in 20% to 40% of patients. Combining drugs with fast
and slow rates of recovery from Na+ channel block (e.g., mexiletine
plus quinidine) can be effective, with fewer adverse effects, when
neither drug alone is effective. Lidocaine and similar agents (mexiletine, tocainide,
phenytoin) with short Na+ Channel Blocker Toxicity Conduction slowing in potential reentrant circuits can account for toxicity due to Na+ channel block (Table 351). For example, Na+ channel block decreases conduction velocity and hence slows atrial flutter rate. Normal AV nodal function permits a greater number of impulses to penetrate the ventricle, and heart rate actually may increase (Figure 359). Thus, atrial flutter rate may drop from 300 per minute, with 2:1 or 4:1 atrioventricular conduction (i.e., a heart rate of 75 or 150 per minute), to 220 per minute, but with 1:1 transmission to the ventricle (i.e., a heart rate of 220 per minute). This form of drug-induced arrhythmia is especially common during treatment with quinidine because the drug also increases AV nodal conduction through its vagolytic properties; flecainide and propafenone also have been implicated. Therapy with Na+ channel blockers in patients with reentrant ventricular tachycardia after a myocardial infarction can increase the frequency and severity of arrhythmic episodes, presumably because conduction slowing allows persistence of the reentrant wavefront within the tachycardia circuit. Such drug-exacerbated arrhythmia can be very difficult to manage, and deaths due to intractable drug-induced ventricular tachycardia have been reported. Some studies suggest Na+ infusion may be beneficial. Several Na+ channel blockers (procainamide, quinidine) have been reported to exacerbate neuromuscular paralysis by d-tubocurarine (see Chapter 9: Agents Acting at the Neuromuscular Junction and Autonomic Ganglia). Action Potential Prolongation Most drugs producing this effect do so by blocking K+
channels, although enhanced inward Na+ current also can prolong
action potentials. Enhanced inward current may underlie QT prolongation (and
arrhythmia suppression) by ibutilide. Block of cardiac K+
channels increases action potential duration and reduces normal automaticity
(Figure 3510 D). Increased action potential duration, seen as an
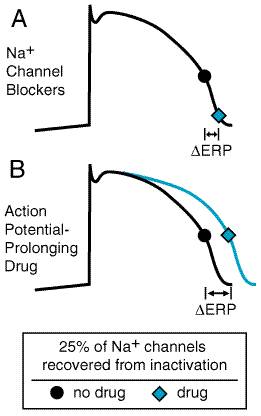
increase in QT interval, increases refractoriness (Figure 3511), which
should be an effective way of treating reentry (Task Force, 1991; Singh, 1993).
Some studies have shown that K+ channel block reduces
heterogeneity of refractoriness, an effect that also should prevent reentrant
arrhythmias. Experimentally, K+ channel block produces a series of
desirable effects: reduced defibrillation energy requirement, inhibition of
ventricular fibrillation due to acute ischemia, and increased contractility (Echt
et al., 1989; Roden, 1993). As shown in Table 353, most K+
channel blocking drugs also interact with Toxicity of Drugs That Prolong QT Interval Most of these agents prolong cardiac action potentials to a disproportionate extent when underlying heart rate is slow; this effect, in turn, results in EADs and related triggered activity in vitro, and can cause torsades de pointes (Table 351; Figure 359). In patients being treated for atrial fibrillation, torsades de pointes occur after conversion to sinus rhythm. For unknown reasons, this form of antiarrhythmic drug toxicity is significantly more common in women (Makkar et al., 1993). Ca2+ Channel Block The major electrophysiological effects resulting from block of cardiac Ca2+ channels are in slow-response tissues, the sinus and AV nodes. Dihydropyridines, such as nifedipine, commonly used in angina and hypertension (Chapters 32: Drugs Used for the Treatment of Myocardial Ischemia and 33: Antihypertensive Agents and the Drug Therapy of Hypertension), preferentially block Ca2+ channels in vascular smooth muscle; their cardiac electrophysiological effects, such as heart rate acceleration, are indirect and attributable to sympathetic activation. Only verapamil, diltiazem, and bepridil block Ca2+ channels in cardiac cells at clinically used doses. With these drugs, heart rate generally is slowed (Figure 3510A), although hypotension, if marked, can cause reflex sympathetic activation and tachycardia. AV nodal conduction velocity decreases, so the PR interval increases. AV nodal block occurs as a result of decremental conduction as well as increased AV nodal refractoriness. These latter effects form the basis of the antiarrhythmic actions of Ca2+ channel blockers in reentrant arrhythmias whose circuit involves the AV node, such as AV reentrant tachycardia (Figure 357). Another important antiarrhythmic action is reduction of ventricular
rate in atrial flutter or fibrillation. Rare forms of ventricular tachycardia
appear to be DAD-mediated and respond to verapamil (Sung et al., 1983).
Unlike Verapamil and Diltiazem The major adverse effect of intravenous verapamil or diltiazem is
hypotension, particularly with bolus doses. This is a particular problem if
the drugs are mistakenly used in patients with ventricular tachycardia (in
which Ca2+ channel blockers are not usually effective)
misdiagnosed as AV nodal reentrant tachycardia (Stewart et al., 1986).
Hypotension also is frequent in patients receiving other vasodilators,
including quinidine, and in patients with underlying left ventricular
dysfunction, which the drugs can exacerbate. Severe sinus bradycardia or
heart block also occurs, especially in susceptible patients, such as those
also receiving Verapamil CALAN ISOPTIN VERELAN COVERA HS) is prescribed as a racemate. l-Verapamil is a more potent calcium channel blocker than is d-verapamil. However, with oral therapy, the l-enantiomer undergoes more extensive first-pass hepatic metabolism. For this reason, a given concentration of verapamil prolongs the PR interval to a greater extent when the drug is administered intravenously (where concentrations of the l- and d-enantiomers are equivalent) than when it is administered orally (Echizen et al., 1985). Diltiazem (CARDIZEM TIAZAC DILACORXR, and others) also undergoes extensive first-pass hepatic metabolism, and both drugs have metabolites that exert Ca2+ channelblocking actions. In clinical practice, adverse effects during therapy with verapamil or diltiazem are determined largely by underlying heart disease and concomitant therapy; plasma concentrations of these agents are not routinely measured during therapy. Both drugs can increase serum digoxin concentration, although the magnitude of this effect is variable; excess slowing of ventricular response in patients with atrial fibrillation can occur. Block of
As with Ca2+ channel blockers and digitalis, a major effect
of Selected It is likely that most |
Principles in the Clinical Use of Antiarrhythmic Drugs
|
Drugs that modify cardiac electrophysiology often have a very narrow margin between the doses required to produce a desired effect and those associated with adverse effects. Moreover, adverse effects from antiarrhythmic drug therapy can include induction of new arrhythmias, with possibly fatal consequences. Nonpharmacological treatments, such as cardiac pacing, electrical defibrillation, or ablation of targeted regions (Morady, 1999), are indicated for some arrhythmias; in other cases no therapy is required even though an arrhythmia is detected. Therefore, the fundamental principles of therapeutics described here must be applied to optimize antiarrhythmic therapy. Identify and Remove Precipitating Factors Factors that commonly precipitate cardiac arrhythmias include hypoxia, electrolyte disturbances (especially hypokalemia), myocardial ischemia, and certain drugs. Antiarrhythmics, including digitalis glycosides, are not the only drugs that can precipitate arrhythmias (Table 351). For example, theophylline is a common cause of multifocal atrial tachycardia, which sometimes can be managed simply by reducing the dose of theophylline. Torsades de pointes can arise not only during therapy with action potential-prolonging antiarrhythmics, but also with other drugs not ordinarily classified as having effects on ion channels. These include the antihistamines terfenadine and astemizole (Chapter 25: Histamine, Bradykinin, and Their Antagonists); the antibiotic, erythromycin (Chapter 47: Antimicrobial Agents: Protein Synthesis Inhibitors and Miscellaneous Antibacterial Agents); the antiprotozoal, pentamidine (Chapter 41: Drugs Used in the Chemotherapy of Protozoal Infections: Amebiasis, Giardiasis, Trichomoniasis, Trypanosomiasis, Leishmaniasis, and Other Protozoal Infections); some antipsychotics, notably thioridazine (Chapter 20: Drugs and the Treatment of Psychiatric Disorders: Psychosis and Mania); and certain tricyclic antidepressants (Chapter 19: Drugs and the Treatment of Psychiatric Disorders: Depression and Anxiety Disorders). Establish the Goals of Treatment Some Arrhythmias Should Not Be Treated: The CAST Example Abnormalities of cardiac rhythm are readily detectable by a variety of
recording methods. However, the mere detection of an abnormality should not
be equated with the need for therapy. This was best illustrated in the
Cardiac Arrhythmias Suppression Trial (CAST). The presence of asymptomatic
ventricular ectopic beats is known to be a marker for an increased risk of
sudden death due to ventricular fibrillation in patients convalescing from a
myocardial infarction. In the CAST, patients in whom ventricular ectopic
beats were suppressed by the potent Na+ channel blockers encainide
(no longer marketed) or flecainide were randomly assigned to receive those
drugs or a matching placebo. Unexpectedly, the mortality rate was two-to
threefold higher among patients treated with the drugs than those treated
with placebo (CAST Investigators, 1989). While the explanation of this effect
is not known, a number of lines of evidence suggest that, in the presence of
these drugs, transient episodes of myocardial ischemia and/or sinus
tachycardia can cause marked conduction slowing (because these drugs have a
very long Symptoms Due to Arrhythmias Some patients with an arrhythmia may be asymptomatic; in this case, establishing any benefit for treatment will be very difficult. Some patients may present with presyncope, syncope, or even cardiac arrest, which may be due to brady- or tachyarrhythmias. Other patients may present with a sensation of irregular heartbeats, which can be minimally symptomatic in some individuals and incapacitating in others. The irregular heartbeats may be due to intermittent premature contractions or to sustained arrhythmias such as atrial fibrillation (which results in an irregular ventricular rate; Figure 359). Finally, patients may present with symptoms due to decreased cardiac output attributable to arrhythmias. The most common symptom is breathlessness at rest or on exertion. Rarely, patients with sustained tachycardias will present with congestive heart failure, which can be controlled by treating the arrhythmia. Choosing Among Therapeutic Approaches Establishing the goals of therapy is especially important when
different therapeutic options are available. For example, in patients with
atrial fibrillation, three options are available: (1) Reduce the ventricular
response, using AV nodal blocking agents such as digitalis, verapamil, diltiazem,
or Factors that contribute to choice of therapy include not only symptoms but also the type and extent of structural heart disease, the QT interval prior to drug therapy, the coexistence of conduction system disease, and the presence of noncardiac diseases (Table 354). In the rare patient with the WPW syndrome and atrial fibrillation, the ventricular response can be extremely rapid and can be paradoxically accelerated by AV nodal blocking drugs such as digitalis or Ca2+ channel blockers; deaths due to drug therapy have been reported under these circumstances. Frequency and reproducibility of arrhythmia should be established prior to initiating therapy, since inherent variability in the occurrence of arrhythmias can be confused with a beneficial or adverse drug effect. Techniques for this assessment include recording cardiac rhythm for prolonged periods or evaluating the response of the heart to artificially induced premature beats. It also is important to recognize that drug therapy may be only partially effective: a marked decrease in the duration of paroxysms of atrial fibrillation may be sufficient to render a patient asymptomatic, even if an occasional episode still can be detected. Minimize Risks Antiarrhythmic Drugs Can Cause Arrhythmias One increasingly well-recognized risk of antiarrhythmic therapy is the possibility of provoking new arrhythmias, with potentially life-threatening consequences. Mechanistically distinct syndromes of arrhythmia provocation by antiarrhythmic drugs have been described (Table 351). These drug-provoked arrhythmias must be recognized, since further treatment with antiarrhythmic drugs often exacerbates the problem, whereas withdrawal of the causative agent often is curative. In addition, specific therapies targeted toward underlying mechanisms of these arrhythmias may be indicated. It also is critical to establish a precise diagnosis. For example, treating a ventricular tachycardia with verapamil not only may be ineffective but also can cause catastrophic cardiovascular collapse (Stewart et al., 1986). Monitoring of Plasma Concentration Some adverse effects of antiarrhythmic drugs are related to excessively elevated plasma drug concentration. Thus, measuring plasma concentration and adjusting the dose to maintain the concentration within a prescribed therapeutic range may be a useful way of minimizing some adverse effects. In many patients, the occurrence of serious adverse reactions appears to be related to interactions involving drug (often at usual plasma concentrations), transient factors such as electrolyte disturbances or myocardial ischemia, and the type and extent of the underlying heart disease (Ruskin, 1989; Morganroth et al., 1986; Roden, 1994). Factors such as generation of unmeasured active metabolites, variability in elimination of enantiomers (which may exert differing pharmacological effects), and disease-or enantiomer-specific abnormalities in drug binding to plasma proteins can complicate the interpretation of routine monitoring of plasma drug concentrations (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination). Patient-Specific Contraindications Another way of minimizing the adverse effects of antiarrhythmic drugs is to avoid certain drugs in certain patient subsets altogether. For example, patients with a history of congestive heart failure are particularly prone to develop heart failure during disopyramide therapy. Often, adverse effects of drugs may be difficult to distinguish from exacerbations of underlying disease. Amiodarone may cause interstitial lung disease; its use is therefore undesirable in a patient with advanced pulmonary disease in whom the development of this potentially fatal adverse effect would be difficult to detect. Specific diseases that constitute relative or absolute contraindications to specific drugs are listed in Table 354. The Electrophysiology of the Heart as a 'Moving Target' Cardiac electrophysiology varies in a highly dynamic fashion in response to external influences such as changing autonomic tone, myocardial ischemia, or myocardial stretch. For example, myocardial ischemia results in changes in extracellular K+ that, in turn, make the resting potential less negative, inactivate Na+ channels, decrease Na+ current, and slow conduction (Weiss et al., 1991). In addition, myocardial ischemia can result in the release of 'metabolites of ischemia,' such as lysophosphatidylcholine, which can alter ion channel function (DaTorre et al., 1991); ischemia also may activate channels that otherwise are quiescent, such as the ATP-inhibited K+ channels (Wilde and Janse, 1994). Thus, a normal heart may display, in response to myocardial ischemia, changes in resting potential, conduction velocity, intracellular Ca2+ concentrations, and repolarization, any one of which may then create arrhythmias or alter response to antiarrhythmic therapy. |
Antiarrhythmic Drugs
|
Summaries of important electrophysiological
and pharmacokinetic features of the drugs considered here are presented in Tables
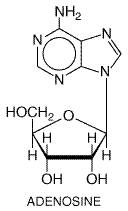
353 and 355. Ca2+ channel blockers and Adenosine Adenosine ADENOCARD) is a naturally occurring nucleoside which is administered as a rapid intravenous bolus for the acute termination of reentrant supraventricular arrhythmias (Lerman and Belardinelli, 1991). Rare cases of ventricular tachycardia in patients with otherwise normal hearts are thought to be DAD-mediated and can be terminated by adenosine. Adenosine also has been used to produce controlled hypotension during some surgical procedures and in the diagnosis of coronary artery disease. Intravenous ATP appears to produce effects similar to those of adenosine.
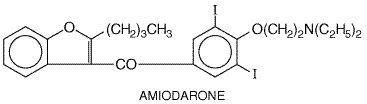
Pharmacological Effects The effects of adenosine are mediated by its interaction with specific G proteincoupled adenosine receptors. Adenosine activates acetylcholine-sensitive K+ current in the atrium and sinus and AV nodes, resulting in shortening of action potential duration, hyperpolarization, and slowing of normal automaticity (Figure 3510C). Adenosine also inhibits the electrophysiological effects of increased intracellular cyclic AMP, which occur with sympathetic stimulation. Because adenosine thereby reduces Ca2+ currents, it can be antiarrhythmic by increasing AV nodal refractoriness and by inhibiting DADs elicited by sympathetic stimulation. Administration of an intravenous bolus of adenosine to human beings transiently slows sinus rate and AV nodal conduction velocity and increases AV nodal refractoriness. It has been shown that a bolus dose of adenosine can produce transient sympathetic activation by interacting with carotid baroreceptors (Biaggioni et al., 1991); when a continuous infusion of the drug is administered, hypotension results. Adverse Effects A major advantage of adenosine therapy is that adverse effects are short-lived, since the drug is eliminated so rapidly. Transient asystole (lack of any cardiac rhythm whatsoever) is common but usually lasts <5 seconds and is in fact the therapeutic goal. Most patients feel a sense of chest fullness and dyspnea when therapeutic doses (6 to 12 mg) of adenosine are administered. Rarely, an adenosine bolus can precipitate atrial fibrillation, presumably by heterogeneously shortening atrial action potentials, or bronchospasm. Clinical Pharmacokinetics Adenosine is eliminated with a half-life of seconds by carrier-mediated uptake, which occurs in most cell types including the endothelium, and subsequent metabolism by adenosine deaminase. Adenosine probably is the only drug whose efficacy requires a rapid bolus dose, preferably through a large central intravenous line; slow administration results in elimination of the drug prior to its arrival at the heart. The effects of adenosine are potentiated in patients receiving dipyridamole, an adenosine-uptake inhibitor, and in patients with cardiac transplants, due to denervation hypersensitivity. Methylxanthines (see Chapter 28: Drugs Used in the Treatment of Asthma) such as theophylline and caffeine block adenosine receptors; therefore larger-than-usual doses are required to produce an antiarrhythmic effect in patients who have consumed these agents in beverages or as therapy. Amiodarone Amiodarone CORDARONE PACERONE) exerts a multiplicity of
pharmacological effects, none of which is clearly linked to its
arrhythmia-suppressing properties (Mason, 1987). Amiodarone is a structural
analog of thyroid hormone, and some of its antiarrhythmic actions and its
toxicity may be attributable to interaction with nuclear thyroid hormone
receptors. Amiodarone is highly lipophilic, is concentrated in many tissues,
and is eliminated extremely slowly; consequently, adverse effects may be very
slow to resolve. In the
Pharmacological Effects Studies of the acute effects of amiodarone in in vitro systems
are complicated by its insolubility in water, necessitating the use of
solvents such as dimethyl sulfoxide. It has been suggested that amiodarone's
effects are mediated by perturbation of the lipid milieu in which ion
channels are placed (Herbette et al., 1988). Amiodarone blocks
inactivated Na+ channels and has a relatively rapid (time constant
Adverse Effects Hypotension due to vasodilation and depression of myocardial performance is frequent with the intravenous form of amiodarone, and may be due in part to the solvent. While depression of contractility can occur during long-term therapy, it is unusual. During oral drug-loading regimens, which usually take place over several weeks, adverse effects are unusual, despite administration of high dosages that would cause serious toxicity if continued long-term. Occasional patients develop nausea, which responds to a decrease in daily dose during the loading phase. Adverse effects during long-term therapy have been related to both the
size of daily maintenance doses as well as to cumulative dose (i.e.,
to duration of therapy), suggesting tissue accumulation may be responsible.
The most serious adverse effect during chronic amiodarone therapy is
pulmonary fibrosis, which can be rapidly progressive and fatal. Underlying
lung disease, doses Clinical Pharmacokinetics Amiodarone is incompletely ( A therapeutic plasma amiodarone concentration range of 0.5 to 2.0 Dose adjustments are not required in conditions such as hepatic, renal, or cardiac dysfunction. Amiodarone is a potent inhibitor of the hepatic metabolism or renal elimination of many compounds. Mechanisms identified to date include inhibition of CYP3A4 and CYP2C9 and of P-glycoprotein (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination). Dosages of warfarin, other antiarrhythmics (flecainide, procainamide, quinidine), or digoxin usually require reduction during amiodarone therapy. Bretylium Bretylium (bretylium tosylate; BRETYLOL) is a quaternary ammonium compound that prolongs cardiac action potentials and interferes with reuptake of norepinephrine by sympathetic neurons; both actions may be antiarrhythmic (Heissenbuttel and Bigger, 1979). Bretylium loading and maintenance infusions are used to treat ventricular fibrillation and prevent its recurrence (see Table 355 for loading and maintenance doses).
Pharmacological Effects Bretylium prolongs action potentials in normal Purkinje cells to a greater extent than in cells that have survived a recent ischemic insult (in which action potentials already are prolonged abnormally). Thus, bretylium reduces heterogeneity of repolarization times, an effect that may suppress reentry (Cardinal and Sasyniuk, 1978). The mechanism whereby bretylium prolongs cardiac action potentials has not been established, although block of K+ channels seems likely. Bretylium has no effect on Na+ channels, except at high concentrations, and no direct effect on automaticity. In animals and human beings, administration of bretylium initially results in increased norepinephrine release from sympathetic neurons and inhibition of subsequent reuptake. Adverse Effects As a result of norepinephrine release, bretylium can produce transient hypertension and increased arrhythmias; this effect rarely is observed, since bretylium is used in critically ill patients who often are hemodynamically unstable. In theory, bretylium should be avoided in patients who are especially prone to increased arrhythmias with norepinephrine release, such as those with digitalis intoxication. In contrast, hypotension due to inhibition of norepinephrine reuptake is a common problem during bretylium therapy. Bretylium-induced hypotension should be managed with judicious fluid replacement if possible. Since bretylium effectively results in sympathetic denervation, the administration of normal doses of catecholamines such as dopamine may cause marked hypertension. Bretylium should be used only with great caution when the drug's vasodilating effects may be particularly hazardous, as in patients with aortic stenosis, carotid occlusive disease, or hypertrophic cardiomyopathy. The occurrence of torsades de pointes is unusual during bretylium therapy. Clinical Pharmacokinetics Bretylium is excreted unchanged by the kidneys without undergoing
significant hepatic metabolism. Reduction of a maintenance infusion rate has
been recommended in patients with renal failure, although adverse effects due
to accumulation of bretylium in plasma have not been seen. A lag time of An oral formulation has been investigated, but hypotension and reduced
bioavailability ( Digitalis Glycosides Pharmacological Effects Digitalis glycosides exert positive inotropic effects and are widely
used in heart failure (Chapter 34: Pharmacological Treatment of Heart Failure).
Their inotropic action is the result of increased intracellular Ca2+
(Smith, 1988), which also forms the basis for arrhythmias related to
digitalis intoxication. Digitalis glycosides increase phase 4 slope (i.e.,
increase the rate of automaticity), especially if [K]o
is low. Digitalis glycosides also exert prominent vagotonic actions,
resulting in inhibition of Ca2+ currents in the AV node and
activation of acetylcholine-mediated K+ currents in the atrium.
Thus, the major 'indirect' electrophysiological effects of
digitalis glycosides are hyperpolarization, shortening of atrial action
potentials, and increases in AV nodal refractoriness. The latter action
accounts for the utility of digitalis in termination of reentrant arrhythmias
involving the AV node, and in controlling ventricular response in patients
with atrial fibrillation. Digitalis preparations may be especially useful in
the latter situation, since many such patients have heart failure, which can
be exacerbated by other AV nodal blocking drugs such as Ca2+
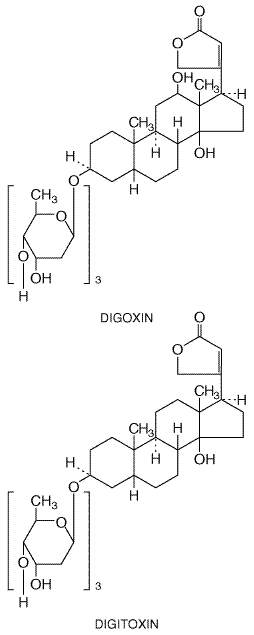
channel blockers or Adverse Effects Digitalis intoxication is a common clinical problem (see also Chapter 34: Pharmacological Treatment of Heart Failure). Arrhythmias, nausea, disturbances of cognitive function, and blurred or yellow vision are the usual manifestations. Often, these are not immediately recognized, since they occur frequently in patients with advanced illnesses receiving multiple drugs. Elevated serum concentrations of digitalis, hypoxia (e.g., due to chronic lung disease), and hypokalemia, hypomagnesemia, and hypercalcemia predispose patients to digitalis-induced arrhythmias. While digitalis intoxication can cause virtually any arrhythmia, certain types of arrhythmias are characteristic. Arrhythmias that should raise a strong suspicion of digitalis intoxication are those in which DAD-related tachycardias occur along with impairment of sinus node or AV nodal function. Atrial tachycardia with AV block is 'classic,' but ventricular bigeminy (sinus beats alternating with beats of ventricular origin), 'bidirectional' ventricular tachycardia (a very rare entity), AV junctional tachycardias, and various degrees of AV block also can occur. With advanced intoxication (e.g., with suicidal ingestion), severe hyperkalemia due to poisoning of Na+,K+ATPase and profound bradyarrhythmias, which may be unresponsive to pacing therapy, are seen. In patients with elevated serum digitalis blood levels, the risk of precipitating ventricular fibrillation by DC cardioversion probably is increased; in those with therapeutic blood levels, DC cardioversion can be used safely. Minor forms of digitalis intoxication may require no specific therapy beyond monitoring cardiac rhythm until symptoms and signs of toxicity resolve. Sinus bradycardia and AV block often respond to intravenous atropine, but the effect is transient. Mg2+ has been used successfully in some cases of digitalis-induced tachycardia (Seller, 1971). Any serious arrhythmia should be treated with antidigoxin Fab fragments, which are highly effective in binding digoxin and digitoxin, greatly enhancing the renal excretion of these drugs (see Chapter 34: Pharmacological Treatment of Heart Failure). Serum glycoside concentrations rise markedly with antidigitalis antibodies, but these represent bound (nonpharmacologically active) drug. Temporary cardiac pacing may be required for advanced sinus node or AV node dysfunction. Other forms of antidotal therapy that have been used in the past, but have been largely supplanted by the use of Fab fragments, include lidocaine, phenytoin, or the cautious administration of K+ for ventricular arrhythmias; K+ can exacerbate AV block. Digitalis exerts direct arterial vasoconstrictor effects, which can be especially deleterious in patients with advanced atherosclerosis who receive intravenous drug; ischemia in the intestinal or coronary beds has been reported. Clinical Pharmacokinetics The most commonly used digitalis glycoside in the
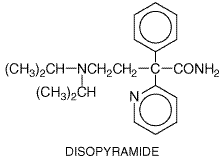
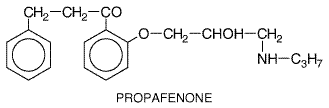
The elimination half-life of digoxin ordinarily is Quinidine elevates serum digoxin concentrations by decreasing clearance and volume of distribution; new steady-state digoxin concentrations are approached in 4 to 5 digoxin elimination half-lives, i.e., in about a week (Leahey et al., 1978). A similar effect has been reported with digitoxin when serum concentrations were monitored for sufficiently long periods. Digitalis toxicity results so often when quinidine is being administered that it is routine to decrease the dose of digoxin if quinidine is started. Other drugs that increase serum digoxin concentration include verapamil, diltiazem, amiodarone, cyclosporine, itraconazole, propafenone, flecainide, and spironolactone. In these cases, the effect is less predictable, and digoxin concentrations regularly are measured and the dose adjusted only if necessary. One common mechanism underlying digoxin clearance appears to involve P-glycoproteinmediated transport. Inhibition of this transport has been implicated as the mechanism whereby quinidine and other interacting drugs decrease digoxin clearance (Fromm et al., 1999). Hypokalemia, which can be caused by many drugs (e.g., diuretics, amphotericin B, corticosteroids), will potentiate digitalis-induced arrhythmias. Disopyramide Disopyramide NORPACE, others; Morady et al., 1982) exerts electrophysiological effects very similar to those of quinidine, but the drugs have different adverse-effect profiles. Disopyramide is used for the maintenance of sinus rhythm in patients with atrial flutter or atrial fibrillation and for the prevention of recurrence of ventricular tachycardia or ventricular fibrillation. Disopyramide is prescribed as a racemate. Its structure is given below.
Pharmacological Actions and Adverse Effects The in vitro electrophysiological actions of S-(+)-disopyramide
are similar to those of quinidine (Mirro et al., 1981). The R-()-enantiomer
produces similar Na+ channel block but does not prolong cardiac
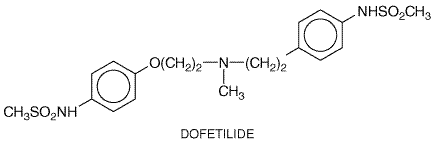
action potentials. Unlike quinidine, racemic disopyramide is not an Clinical Pharmacokinetics Disopyramide is well absorbed. Binding to plasma proteins is concentration-dependent, so a small increase in total concentration may represent a proportionately larger increase in free drug concentration (Lima et al., 1981). Disopyramide is eliminated by both hepatic metabolism (to a weakly active metabolite) and renal excretion of unchanged drug. The dose should be reduced in patients with renal dysfunction. Higher-than-usual dosages may be required in patients receiving drugs that induce hepatic metabolism, such as phenytoin. Dofetilide Dofetilide(TIKOSYN) is a potent and 'pure'IKr blocker. As a result of this specificity, it has virtually no extracardiac pharmacological effects. Dofetilide is effective in maintaining sinus rhythm in patients with atrial fibrillation. In the DIAMOND studies (Torp-Pedersen et al., 1999), dofetilide did not affect mortality in patients with advanced heart failure or in those convalescing from acute myocardial infarction. Dofetilide currently is available through a restricted distribution system that includes only those physicians, hospitals, and other institutions that have received special educational programs covering proper dosing and treatment initiation.
Adverse Effects Torsades de pointes occurred in 1% to 3% of patients in clinical trials, in whom strict exclusion criteria (e.g., hypokalemia) were applied and continuous ECG monitoring was used to detect marked QT prolongation in the hospital. The incidence of this adverse effect during more widespread use of the drug, marketed in 2000, is unknown. Other adverse effects are no more common than with placebo. Clinical Pharmacokinetics Most of a dose of dofetilide is excreted unchanged by the kidneys. In patients with mild to moderate renal failure, decreases in dosage based on creatinine clearance are required to minimize the risk of torsades de pointes. The drug should not be used in patients with advanced renal failure and should not be used with inhibitors of renal cation transport. Dofetilide also undergoes minor hepatic metabolism. Flecainide The effects of flecainide (TAMBOCOR) therapy are thought to be
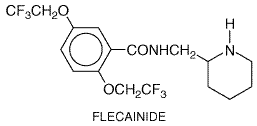
attributable to the drug's very long
Pharmacological Effects Flecainide blocks Na+ current and delayed rectifier K+
current (IKr) at similar concentrations in vitro, 1
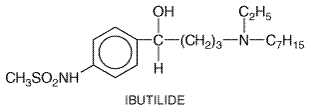
to 2 Adverse Effects Flecainide produces few subjective complaints in most patients; dose-related blurred vision is the most common noncardiac adverse effect. It can exacerbate congestive heart failure in patients with depressed left ventricular performance. The most serious adverse effects are provocation or exacerbation of potentially lethal arrhythmias. These include acceleration of ventricular rate in patients with atrial flutter, increased frequency of episodes of reentrant ventricular tachycardia, and increased mortality in patients convalescing from myocardial infarction (Morganroth et al., 1986; Crijns et al., 1988; CAST Investigators, 1989; Ranger et al., 1989). As discussed above, it is likely that all these effects can be attributed to Na+ channel block. Flecainide also can cause heart block in patients with conduction system disease. Clinical Pharmacokinetics Flecainide is well absorbed. The elimination half-life is shorter with urinary acidification (10 hours) than with urinary alkalinization (17 hours), but it is nevertheless sufficiently long to allow dosing twice daily (see Table 355). Elimination occurs by both renal excretion of unchanged drug and hepatic metabolism to inactive metabolites. The latter is mediated by the polymorphically distributed enzyme CYP2D6 (Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination) (Gross et al., 1989). However, even in patients in whom this pathway is absent because of genetic polymorphism or inhibition by other drugs (i.e., quinidine, fluoxetine), renal excretion ordinarily is sufficient to prevent drug accumulation. In the rare patient with renal dysfunction and lack of active CYP2D6, flecainide may accumulate to toxic plasma concentrations. Flecainide is a racemate, but there are no differences in the electrophysiological effects or disposition kinetics of its enantiomers (Kroemer et al., 1989). Some reports have suggested that plasma flecainide concentrations >1000 ng/ml should be avoided to minimize the risk of flecainide toxicity; however, in susceptible patients, the adverse electrophysiological effects of flecainide therapy can occur at therapeutic plasma concentrations. Ibutilide Ibutilide CORVERT; Murray, 1998) is an IKr blocker that in some systems also activates an inward Na+ current. The action potential-prolonging effect of the drug may arise from either mechanism. Ibutilide is administered as a rapid infusion (1 mg over 10 minutes) for the immediate conversion of atrial fibrillation or flutter to sinus rhythm. The drug's efficacy rate is higher in patients with atrial flutter (50% to 70%) than in those with atrial fibrillation (30% to 50%). In atrial fibrillation, the conversion rate is lowest in those in whom the arrhythmia has been present for weeks or months compared with those in whom it has been present for days. The major toxicity with ibutilide is torsades de pointes, which occur in up to 6% of patients and requires immediate cardioversion in up to one-third of these. The drug undergoes extensive first-pass metabolism and so is not used orally. It is eliminated by hepatic metabolism and has a half-life of 2 to 12 hours (average of 6 hours).
Lidocaine Lidocaine XYLOCAINE) is a local anesthetic that also is useful in the acute intravenous therapy of ventricular arrhythmias. When lidocaine was administered to all patients with suspected myocardial infarction, the incidence of ventricular fibrillation was reduced (Lie et al., 1974). However, survival to hospital discharge tended to be decreased (Hine et al., 1989), perhaps because of lidocaine-exacerbated heart block or congestive heart failure. Lidocaine is therefore no longer routinely administered to all patients in coronary care units.
Pharmacological Effects Lidocaine blocks both open and inactivated cardiac Na+ channels. Findings from in vitro studies suggest that lidocaine-induced block reflects an increased likelihood that the Na+ channel protein assumes a nonconducting conformation in the presence of drug (Balser et al., 1996). Recovery from block is very rapid, so lidocaine exerts greater effects in depolarized (e.g., ischemic) and/or rapidly driven tissues. Lidocaine is not useful in atrial arrhythmias, possibly because atrial action potentials are so short that the Na+ channel is in the inactivated state only briefly compared with diastolic (recovery) times, which are relatively long (Hondeghem and Katzung, 1984). In some studies, lidocaine increased current through inward rectifier channels, but the clinical significance of this effect is not known. Lidocaine can hyperpolarize Purkinje fibers depolarized by low [K]o or stretch (Arnsdorf and Bigger, 1972); the resultant increased conduction velocity may be antiarrhythmic in reentry. Lidocaine decreases automaticity by reducing the slope of phase 4 and altering the threshold for excitability. Action potential duration usually is unaffected or is shortened; such shortening may be due to block of the few Na+ channels that inactivate late during the cardiac action potential. Lidocaine usually exerts no significant effect on PR or QRS duration; QT is unaltered or slightly shortened. The drug exerts little effect on hemodynamic function, although rare cases of lidocaine-associated exacerbations of heart failure have been reported, especially in patients with very poor left ventricular function. Adverse Effects When a large intravenous dose of lidocaine is administered rapidly, seizures can occur. When plasma concentrations of the drug rise slowly above the therapeutic range, as may occur during maintenance therapy, tremor, dysarthria, and altered levels of consciousness are more common. Nystagmus is an early sign of lidocaine toxicity. Clinical Pharmacokinetics Lidocaine is well absorbed, but undergoes extensive, though variable, first-pass hepatic metabolism (Thompson et al., 1973); thus, oral use of the drug is inappropriate. In theory, therapeutic plasma concentrations of lidocaine may be maintained by intermittent intramuscular administration, but the intravenous route is preferred (see Table 355). Lidocaine's metabolites, glycine xylidide (GX) and mono-ethyl GX (MEGX), are less potent as Na+ channel blockers than the parent drug. GX and lidocaine appear to compete for access to the Na+ channel, suggesting that, with infusions during which GX accumulates, lidocaine's efficacy may be diminished (Bennett et al., 1988). With infusions lasting longer than 24 hours, the clearance of lidocaine fallsan effect that has been attributed to competition between parent drug and metabolites for access to hepatic drug-metabolizing enzymes (LeLorier et al., 1977; Suzuki et al., 1984). Plasma concentrations of lidocaine decline biexponentially after a
single intravenous dose, indicating that a multicompartment model (Chapter 1:
Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and
Elimination) is necessary to analyze lidocaine disposition. The initial drop
in plasma lidocaine following intravenous administration occurs rapidly, with
a half-life of In heart failure, the central volume of distribution is decreased, so
the total loading dose should be decreased (Thompson et al., 1973).
Since lidocaine clearance also is decreased, the rate of the maintenance
infusion should be decreased. Lidocaine clearance is also reduced in hepatic
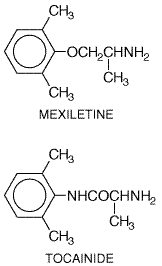
disease (Thompson et al., 1973), during treatment with cimetidine or Magnesium The intravenous administration of 1 to 2 g of MgSO4 has been reported to be effective in preventing recurrent episodes of torsades de pointes, even if serum Mg2+ is normal (Tzivoni et al., 1988). However, controlled studies of this effect have not been performed. The mechanism of action is unknown; since QT interval is not shortened, an effect on the inward current, possibly a Ca2+ current, responsible for the triggered upstroke arising from EADs (black arrow, Figure 356B) is possible (Jackman et al., 1988; Roden, 1991b). Intravenous Mg2+ also has been used successfully in arrhythmias related to digitalis intoxication (Seller, 1971). Large, placebo-controlled trials of intravenous magnesium to improve outcome in acute myocardial infarction have yielded conflicting results (Woods and Fletcher, 1994; ISIS-4 Collaborative Group, 1995). While oral Mg2+ supplements may be useful in preventing hypomagnesemia, there is no evidence that chronic Mg2+ ingestion exerts a direct antiarrhythmic action. Mexiletine and Tocainide Mexiletine MEXITIL) and tocainide (TONOCARD) are analogs of lidocaine with structures that have been modified to reduce first-pass hepatic metabolism to make chronic oral therapy effective (Roden and Woosley, 1986b; Campbell, 1987). Their electrophysiological actions are similar to those of lidocaine. Tremor and nausea are the major dose-related adverse effects; these can be minimized by taking the drugs with food. Because tocainide can cause potentially fatal bone marrow aplasia and pulmonary fibrosis, it is rarely used.
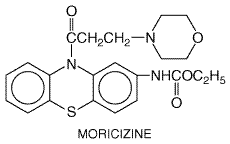
Mexiletine undergoes hepatic metabolism, which is inducible by drugs such as phenytoin. Tocainide, on the other hand, is eliminated by renal excretion. Thus, in patients with renal disease, the dose of tocainide should be decreased. Both agents have been used for ventricular arrhythmias; combinations of mexiletine or tocainide with quinidine or sotalol may increase efficacy while reducing adverse effects. In vitro studies and clinical anecdotes have suggested a role for mexiletine (or flecainide) in correcting the molecular defect in the form of congenital long QT syndrome caused by abnormal Na+ channel inactivation (Shimizu and Antzelevitch, 1997). Moricizine Moricizine ETHMOZINE) is a phenothiazine analog with Na+ channelblocking properties used in the chronic treatment of ventricular arrhythmias (Clyne et al., 1992). In a randomized, double-blind trial (CAST II), moricizine increased mortality in patients shortly after a myocardial infarction and did not improve survival during long-term therapy (Cardiac Arrhythmia Suppression Trial II Investigators, 1992). Moricizine undergoes extensive first-pass hepatic metabolism; despite its short elimination half-life, its antiarrhythmic effect can persist for many hours after a single dose, suggesting that some of its metabolites may be active.
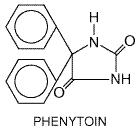
Phenytoin The anticonvulsant phenytoin (DILANTIN; Chapter 21: Drugs Effective in
the Therapy of the Epilepsies) also is a blocker of inactivated cardiac Na+
channels. It has been used in the acute and chronic suppression of
ventricular arrhythmias and in digitalis intoxication (Atkinson and Davison,
1974). Phenytoin has a short
Procainamide Procainamide PROCAN
SR; others) is an
analog of the local anesthetic, procaine. It exerts electrophysiological
effects similar to those of quinidine, but lacks quinidine's vagolytic and
Pharmacological Effects Procainamide is a blocker of open Na+ channels, with an intermediate time constant of recovery from block. It also prolongs cardiac action potentials in most tissues, probably by blocking outward K+ current(s). Procainamide decreases automaticity, increases refractory periods, and slows conduction. The major metabolite, N-acetyl procainamide, lacks the Na+ channelblocking activity of the parent drug but is equipotent in prolonging action potentials (Dangman and Hoffman, 1981). As the plasma concentrations of N-acetyl procainamide often exceed those of procainamide, increased refractoriness and QT prolongation during chronic procainamide therapy can be attributed at least in part to the metabolite. However, it is the parent drug that slows conduction and produces QRS interval prolongation. Although hypotension may occur at high plasma concentrations, this effect usually is attributable to ganglionic blockade rather than to any negative inotropic effect, which is minimal. Adverse Effects Hypotension and marked slowing of conduction are major adverse effects
of high concentrations (>10 During long-term therapy, most patients will develop biochemical evidence of the drug-induced lupus syndrome, such as circulating antinuclear antibodies (Woosley et al., 1978). Therapy need not be interrupted merely because of the presence of antinuclear antibodies. However, many patients, perhaps as many as 25% to 50%, eventually will develop symptoms of the lupus syndrome; common early symptoms are rash and small-joint arthralgias. Other symptoms of lupus, including pericarditis with tamponade, can occur, although renal involvement is unusual. Clinical Pharmacokinetics Procainamide is rapidly eliminated (t1/2= 3 to 4 hours) by both renal excretion of unchanged drug as well as by hepatic metabolism. The major pathway for hepatic metabolism is conjugation by N-acetyl transferase (see Chapter 1: Pharmacokinetics: The Dynamics of Drug Absorption, Distribution, and Elimination) to form N-acetyl procainamide. N-Acetyl procainamide is eliminated by renal excretion (t1/2= 6 to 10 hours) and is not significantly converted back to procainamide. Because of the relatively rapid elimination rates of both the parent drug and its major metabolite, procainamide usually is administered as a slow-release formulation. In patients with renal failure, procainamide and/or N-acetyl procainamide can accumulate to potentially toxic plasma concentrations (Drayer et al., 1977). Reduction of procainamide dose and dosing frequency and monitoring of plasma concentrations of both compounds are required in this situation. Because the parent drug and metabolite exert different pharmacological effects, the practice of using the sum of their concentrations to guide therapy is inappropriate. In individuals who are slow acetylators, the procainamide-induced lupus syndrome develops more often and earlier during treatment than among rapid acetylators (Woosley et al., 1978). In addition, the symptoms of procainamide-induced lupus resolve during treatment with N-acetyl procainamide. Both of these findings support results of in vitro studies suggesting that it is chronic exposure to the parent drug (or an oxidative metabolite) that results in the lupus syndrome; these findings also provided one rationale for the further development of N-acetyl procainamide and its analogs as antiarrhythmic agents (Roden, 1993). Propafenone Propafenone RYTHMOL) is a Na+ channel
blocker with a relatively slow time constant for recovery from block (Funck-Brentano
et al., 1990). Some data suggest that, like flecainide, propafenone also
blocks K+ channels. Its major electrophysiological effect is to
slow conduction in fast-response tissues. The drug is prescribed as a
racemate; while the enantiomers do not differ in their Na+
channelblocking properties, S-(+)-propafenone is a
Adverse effects during propafenone therapy include acceleration of
ventricular response in patients with atrial flutter, increased frequency or
severity of episodes of reentrant ventricular tachycardia, exacerbation of
heart failure, and the adverse effects of Clinical Pharmacokinetics Propafenone is well absorbed and is eliminated by both hepatic and
renal routes. The activity of cytochrome P450 2D6 (CYP2D6), an enzyme that
functionally is absent in Quinidine As early as the eighteenth century, the bark of the cinchona plant was used to treat 'rebellious palpitations' (Levy and Azoulay, 1994). Studies in the early twentieth century identified quinidine, a diastereomer of the antimalarial quinine, as the most potent of the antiarrhythmic substances extracted from the cinchona plant, and by the 1920s quinidine was used as an antiarrhythmic agent (Wenckebach, 1923). Quinidine is used for the maintenance of sinus rhythm in patients with atrial flutter or atrial fibrillation and in the prevention of recurrence of ventricular tachycardia or ventricular fibrillation (Grace and Camm, 1998).
Pharmacological Effects Quinidine (QUINAGLUTE QUINIDEX, others) blocks Na+
current and multiple cardiac K+ currents. It is an open-state
blocker of Na+ channels, with a time constant of recovery in the
intermediate ( Quinidine's Na+ channel-blocking properties result in an increased threshold for excitability and decreased automaticity. As a consequence of its K+ channelblocking actions, quinidine prolongs action potentials in most cardiac cells. This effect is most prominent at slow rates. In some cells, such as midmyocardial cells and Purkinje cells, quinidine consistently elicits EADs at slow heart rates, particularly when [K]o is low (Roden and Hoffman, 1985). Quinidine prolongs refractoriness in most tissues, probably as a result of both prolongation of action potential duration and its Na+ channel blockade. In the intact animal and in human beings, quinidine also produces Adverse Effects Noncardiac Diarrhea is the most common adverse effect during quinidine therapy, occurring in 30% to 50% of patients. The mechanism is not known. Diarrhea usually occurs within the first several days of quinidine therapy, but can occur later. In mild cases of diarrhea in which quinidine therapy is deemed vital, antidiarrheal drugs can be used. Diarrhea-induced hypokalemia may potentiate torsades de pointes due to quinidine. A number of immunological reactions can occur during quinidine therapy. The most common is thrombocytopenia, which can be severe but which resolves rapidly with discontinuation of the drug. Hepatitis, bone marrow depression, and a lupus syndrome occur rarely. None of these effects is related to elevated plasma quinidine concentrations. Quinidine also can produce cinchonism, a symptom complex that includes headache and tinnitus. In contrast to other adverse events during quinidine therapy, cinchonism usually is related to elevated plasma quinidine concentrations, and can be managed by dose reduction. Cardiac It is estimated that 2% to 8% of patients who receive quinidine therapy will develop marked QT-interval prolongation and torsades de pointes. In contrast to effects of sotalol, N-acetyl procainamide, and many other drugs, quinidine-associated torsades de pointes generally occur at therapeutic, or even subtherapeutic, plasma concentrations (Jackman et al., 1988; Roden, 1991). The reasons for individual susceptibility to this adverse effect are not known. At high plasma concentrations of quinidine, marked Na+ channel block can occur, with resultant ventricular tachycardia. This adverse effect formerly occurred when very high doses of quinidine were used to try to convert atrial fibrillation to normal rhythm, but this aggressive approach to quinidine dosing has now been abandoned, and quinidine-induced ventricular tachycardia due to excess Na+ channel block is unusual. Quinidine can exacerbate heart failure or conduction system disease. However, in most patients with congestive heart failure, quinidine is well tolerated, perhaps because of its vasodilating actions. Clinical Pharmacokinetics Quinidine is well absorbed and is 80% bound to plasma proteins
including albumin and, like lidocaine, the acute phase reactant, There is substantial individual variability in the range of dosages
required to achieve therapeutic plasma concentrations of 2 to 5 Drug Interactions Quinidine is a potent inhibitor of CYP2D6. As a result, the
administration of quinidine to patients receiving drugs that undergo
extensive CYP2D6-mediated metabolism may result in altered drug effects due
to accumulation of parent drug and failure of metabolite formation. For example,
inhibition of CYP2D6-mediated metabolism of codeine to its active metabolite,
morphine, results in decreased analgesia. On the other hand, inhibition of
CYP2D6-mediated metabolism of propafenone results in elevated plasma
propafenone concentrations and increased Quinidine metabolism is inducible by drugs such as phenobarbital or phenytoin (Data et al., 1976). In patients receiving these agents, very high doses of quinidine may be required to achieve therapeutic concentrations. It is important to note that, if therapy with the inducing agent is then stopped, quinidine concentrations may rise to very high levels, and its dosage must be adjusted downward. Cimetidine and verapamil elevate plasma quinidine concentrations, but these effects usually are modest. Sotalol Sotalol BETAPACE) is a nonselective
In the Sotalol prolongs action potential duration throughout the heart and QT
interval on the ECG. It decreases automaticity, slows AV nodal conduction,
and prolongs AV refractoriness both by K+ channel block and block
of |
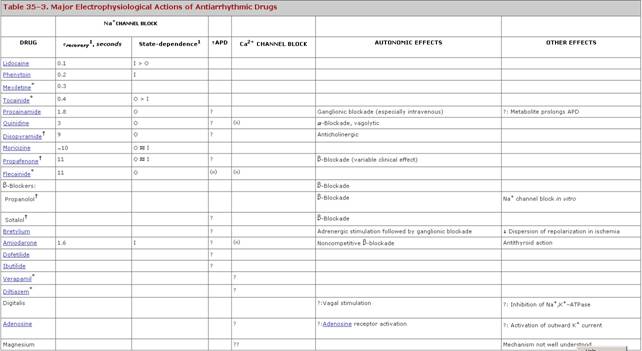
Prospectus
|
Recent and continuing elucidation of the
mechanisms underlying normal and abnormal cardiac electrical behavior, along
with clinical investigations clearly delineating the dangers of
indiscriminate use of potent ion channelblocking drugs in large groups of
patients, have significantly altered therapeutic strategies for the treatment
of arrhythmias. In some instances, nonpharmacological therapies are
increasingly used to avoid the dangers of chronic drug therapy. Techniques
for ablation of areas critical for the genesis of arrhythmias are now
sufficiently well developed that chronic drug therapy can be avoided in many
patients. When arrhythmia recurrence would be lethal and drugs may be
incompletely effective, as in patients resuscitated from an episode of
ventricular fibrillation, implanted defibrillators often are an appropriate
choice. Among drugs discussed here, only For further discussion of cardiac arrhythmias, see Chapters 213
and 214 in Harrison's Principles of Internal Medicine, 16th ed., |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 105965
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2025 . All rights reserved
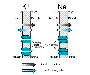
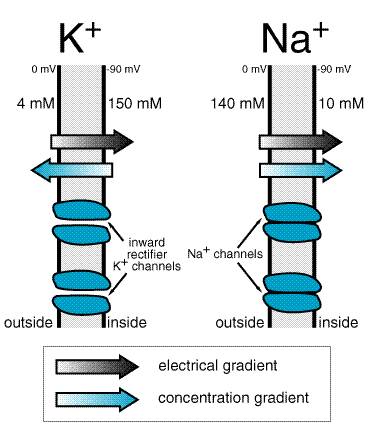


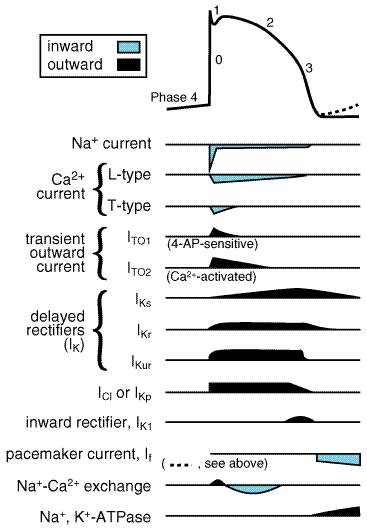

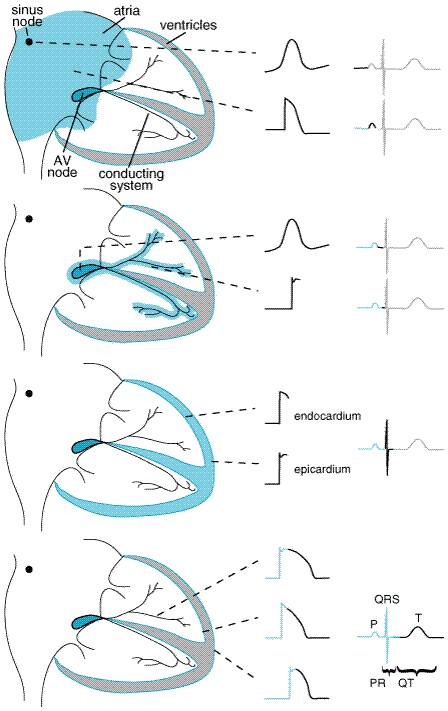
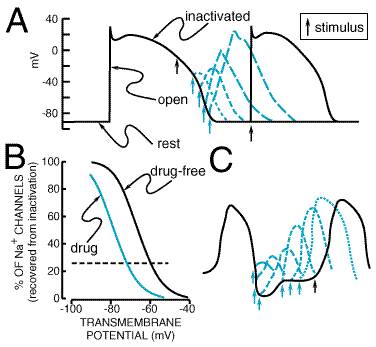
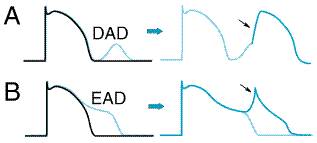
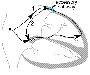

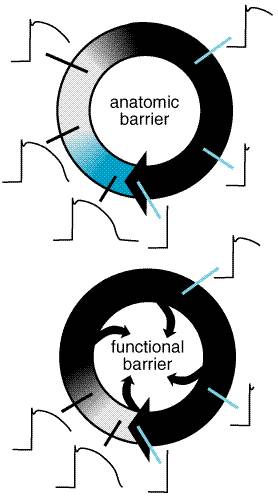
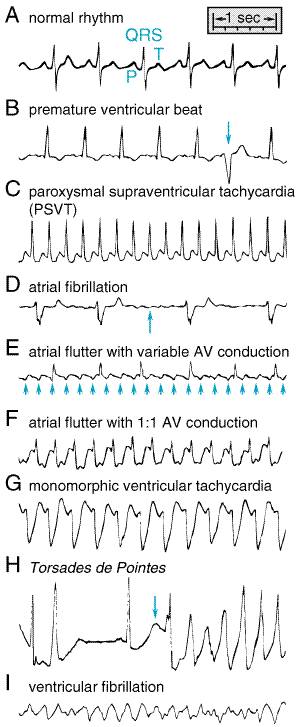 for
defibrillating shocks.
for
defibrillating shocks.