| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Site-directed mutagenesis
Mutagenesis is a fundamentally important DNA technology, which seeks to change the base sequence of DNA and test its effect on gene or DNA function. The mutagenesis can be conducted in vivo (in studies of model organisms, or cultured cells) or in vitro and the mutagenesis can be directed to a specific site in a pre-determined way (site-directed mutagenesis), or can be random. In the case of in vivo mutagenesis, for example, gene targeting offers exquisite site-directed mutagenesis within living cells while exposure of male mice to high levels of a powerful mutagen such as ethyl nitrosurea (ENU) and subsequent mating of the mice offers a form of random mutagenesis which can be important in generating new mutants.
Site-directed mutagenesis production of a specific predetermined change in a DNA sequence. Can be done in vitro on cloned DNA or in vivo by homologous recombination.
In vitro mutagenesis can involve essentially random approaches to mutagenesis, which may be valuable in producing libraries of new mutants. In addition, if a gene has been cloned and a functional assay of the product is available, it is also very useful to be able to employ a form of in vitro mutagenesis, which results in alteration of a specific amino acid or small component of the gene product in a predetermined way.
Many in vitro assays of gene function wish to gain information on the importance of individual amino acids in the encoded polypeptide. This may be relevant when attempting to assess whether a particular missense mutation found in a known disease gene is pathogenic, or just generally in trying to evaluate the contribution of a specific amino acid to the biological function of a protein. In vitro site-directed mutagenesis is an invaluable technique for studying protein structure-function relationships, gene expression and vector modification. Several methods have appeared in the literature, but many of these methods require single-stranded DNA as the template.
Oligonucleotide mismatch mutagenesis is a popular method of introducing a predetermined single nucleotide change into a cloned gene. This method was first developed by Michael Smith who was awarded a Nobel Prize in 1993 for this contribution. Site-directed mutagenesis can also be achieved by using PCR (more info). This approach involves cloning the gene or cDNA into an M13 or phagemid vector, which permits recovery of single-stranded recombinant DNA.
M13 is one of a group of filamentous bacteriophages (including the fd and f1 phages), which can infect certain strains of E. coli. The genomes of these phages consist of single-stranded circular DNA molecules about 6.4 kb long, which are very highly related to each other in DNA sequence and enclosed in a protein coat, forming a long filamentous structure. After adsorption to the bacterium, the phage genome enters the bacterial cell where it is converted from the single-stranded form to a double-stranded form, the replicative form (RF). The latter serves as a template for making numerous copies of the genome and, after a certain time, a phage-encoded product switches DNA synthesis towards production of single strands. The latter migrate to the cell membrane where they are enclosed in a protein coat, and hundreds of mature phage particles are extruded from the infected cell without cell lysis. The cell doesn't die - it just grows more slowly and continues to secrete phage indefinitely. M13 vectors are based upon the double-stranded RF form and have a multiple cloning site for generating double-stranded recombinant DNA circles. The latter can be transfected into suitable strains of E. coli. After a certain period, phage particles are harvested and stripped of their protein coats to release single-stranded recombinant DNA for direct use as templates in DNA sequencing reactions.
Phagemid vectors are plasmids, which have been artificially manipulated so as to contain a small segment of the genome of a filamentous phage, such as M13, fd or f1. The selected phage sequences contain all the cis-acting elements required for DNA replication and assembly into phage particles. They permit successful cloning of inserts several kilobases long (unlike M13 vectors in which such inserts tend to be unstable). Following transformation of a suitable E. coli strain with a recombinant phagemid, the bacterial cells are superinfected with a filamentous helper phage, such as f1, which is required to provide the coat protein. Phage particles secreted from the superinfected cells will be a mixture of helper phage and recombinant phagemids. The mixed single-stranded DNA population can be used directly for DNA sequencing because the primer for initiating DNA strand synthesis is designed to bind specifically to a sequence of the phagemid vector adjacent to the cloning site. Commonly used phagemid vectors include the pEMBL series of plasmids and the pBluescript family.
A mutagenic oligonucleotide primer is then designed whose sequence is perfectly complementary to the gene sequence in the region to be mutated, but with a single difference: at the intended mutation site it bears a base that is complementary to the desired mutant nucleotide rather than the original. The mutagenic oligonucleotide is then allowed to prime new DNA synthesis to create a complementary full-length sequence containing the desired mutation. The newly formed heteroduplex is used to transform cells, and the desired mutant genes can be identified by screening for the mutation
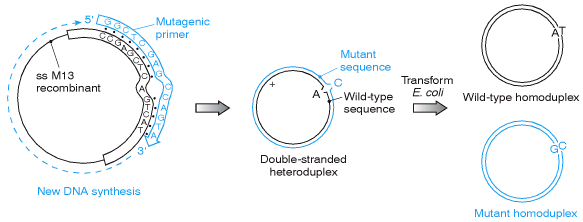
Oligonucleotide mismatch mutagenesis can create a desired point mutation at a unique predetermined site within a cloned DNA molecule. The figure illustrates only one of many different methods of cell-based oligonucleotide mismatch mutagenesis. The example illustrates the use of a mutagenic oligonucleotide to direct a single nucleotide substitution in a gene. The gene is cloned into M13 in order to generate a single-stranded recombinant DNA. An oligonucleotide primer is designed to be complementary in sequence to a portion of the gene sequence encompassing the nucleotide to be mutated (A) and containing the desired noncomplementary base at that position (C, not T). Despite the internal mismatch, annealing of the mutagenic primer is possible, and DNA polymerase can extend second strand synthesis and the gap sealed by DNA ligase. The resulting heteroduplex can be transformed into E. coli, whereupon two populations of recombinants can be recovered: wild type and mutant homoduplexes.
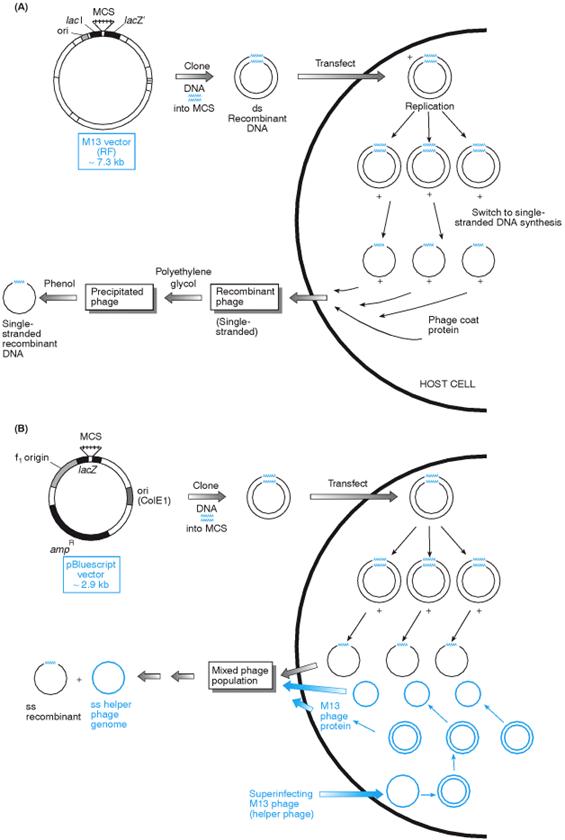
Producing single-stranded recombinant DNA using M13 and phagemid vectors. (A) M13 vectors. M13 vectors are replicative (RF) forms of M13 derivatives containing a nonfunctional component of the lacZ b-galactosidase system, which can be complemented in function by the presence of a complementary lacZ component in the E. coli JM series. The double-stranded M13 recombinant DNA enters the normal cycle of DNA replication to generate numerous copies of the genome, prior to a switch to production of single-stranded DNA (+ strand only). Mature recombinant phage exit from the cell without lysis. (B) Phagemid vectors. The pBluescript series of plasmid vectors contain two origins of replication: a normal one from ColE1 and a second from phage f1, which, in the presence of a filamentous phage genome, will specify production of, single-stranded DNA. Superinfection of transformed cells with M13 phage results in two types of phage-like particles released from the cells: the original superinfecting phage and the plasmid recombinants within a phage protein coat. Sequencing primers specific for the phagemid vector are used to obtain unambiguous sequences.
Approaches that allow us to distinguish one strand from another
Restriction enzymes used to distinguish strands
1. Suppose you had a parental DNA that had a unique restriction site in it. If you mutagenized the restriction site at the same time that you made a mutation in your gene of interest, then the parental strand would be sensitive to the enzyme and the other strand containing the mutation would not. The company Clontech has such a method, called the Transformer Site-Directed Mutagenesis Kit. Digestion of the heteroduplex with the restriction enzyme debilitates the parental strand, because it introduces a 'nick'. The DNA can then be transformed into a bacterial strain. The efficiency can be increased by extracting the pooled DNA from these cells and digesting a second time. This will eliminate the products of replication in the bacteria that are purely parental (homoduplex), and will spare the ones that are purely mutagenized (homoduplex). These plasmids can then be reintroduced into bacteria, and most of the surviving plasmids should be the mutagenized form.
2.There is a restriction enzyme named Dpn I that will cleave the sequence GMeATC where MeA means that the adenylate nucleotide is methylated. Dpn I will not cleave the unmethylated sequence GATC. We can methylate such sequences in a plasmid by growing the plasmid in a 'dam+' strain of bacteria. Suppose then that we prepare a single stranded DNA template in such a 'dam+' strain. The parental strand would be methylated at every GATC sequence (that is, approximately every 200 to 300 nt). When we apply an oligonucleotide primer to this template and extend it using Klenow fragment, however, the new DNA that is synthesized in vitro will be unmethylated. We therefore create a marked difference between the parental strand (methylated) and the mutagenized strand (unmethylated). Once we have completed synthesis of the mutagenized strand, what would happen if we tried to digest the heteroduplex with Dpn I? The answer is that the parental strand would be nicked (cleaved) in many places, but the mutagenized strand would not. By putting this extra damage into the parental strand, it is less favored during replication in the bacteria.
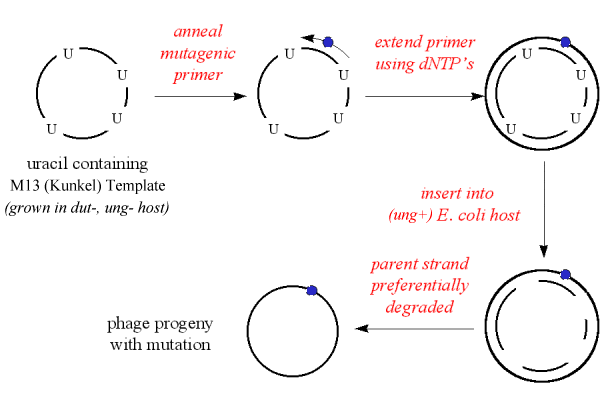
'Kunkel' method
The M13mp vector with insert is first grown in a mutant E. coli host (e.g. CJ236), which would occasionally incorporate uracil into the DNA instead of thymidine.
The Altered States method (Promega kit)
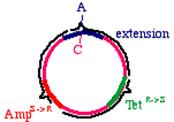
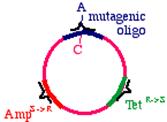
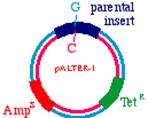
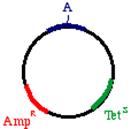
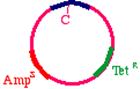
Start with a double stranded plasmid containing your DNA insert, the sequence you wish to mutagenize:
Note that there is a G/C base pair that we want to mutagenize to an A/T base pair, in our dark blue sequence (the parental insert). Also, there is a green sequence representing tetracycline resistance, and a red sequence that is a defective version of the ampicillin resistance gene. Since the Amp gene is defective, we will say that it is AmpS meaning 'sensitive'.
Now we prepare a single stranded version of the plasmid, perhaps by simply denaturing them in alkali. We anneal THREE oligonucleotides to the circle: One to the DNA parental insert, that causes the mutation in our gene of interest (from a G to an A in this example), one to the tetracycline resistance gene that will debilitate it by the introduction of a mutation, and one to the ampicillin 'sensitive' gene that will repair it by the introduction of a mutation.
These oligonucleotides are extended clockwise around the plasmid using DNA polymerase Klenow fragment. Note that this heteroduplex has three points of mismatch, in three entirely different places in the plasmid. The 'inner strand' that contains the parental sequence is unchanged, but the outer strand will contain the three alterations. What happens when the bacteria replicates this? The answer is that two types of products will appear. First, the replicative products of the inner strand. These will carry an intact tetracycline resistance gene and a nonfunctional ampicillin resistance gene. The cells that inherit these plasmids will die in ampicillin. On the other hand, the products of the outer strand. These will carry a functional ampicillin resistance gene, a nonfunctional tetracycline resistance gene, and more importantly, the G->A mutation in the DNA insert.
PCR site-directed methods allow site-specific mutations to be incorporated in virtually any double-stranded plasmid; eliminating the need for M13-based vectors or single-stranded rescue.
Several points should be mentioned concerning site-directed mutagenesis using PCR. First, it is often desirable to reduce the number of cycles during PCR when performing PCR-based site-directed mutagenesis to prevent clonal expansion of any (undesired) second-site mutations. Limited cycling, which would result in reduced product yield, is offset by increasing the starting template concentration. Second, a selection must be used to reduce the number of parental molecules coming through the reaction. Third, in order to use a single PCR primer set, it is desirable to optimize the long PCR method. And fourth, because of the extendase activity of some thermostable polymerases it is often necessary to incorporate an end-polishing step into the procedure prior to end-to-end ligation of the PCR-generated product containing the incorporated mutations in one or both PCR primers.
The 'megaprimer' method of site-directed mutagenesis uses three oligonucleotide primers and two rounds of polymerase chain reaction (PCR). One oligonucleotide is mutagenic; the others are forward and reverse primers that lie upstream and downstream from the binding site for the mutagenic oligonucleotide. The mutagenic primer and the nearer of the external primers are used in the first PCR to generate and amplify a mutated fragment of DNA. This amplified fragmentthe megaprimeris used in the second PCR in conjunction with the remaining external primer to amplify a longer region of the template DNA. This protocol is based on a method that uses forward and reverse external primers with significantly different melting temperatures (Tm
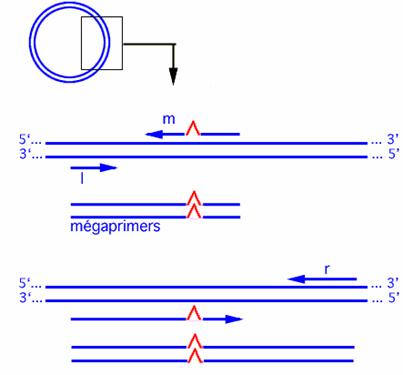
Fig.3 The megaprimer method
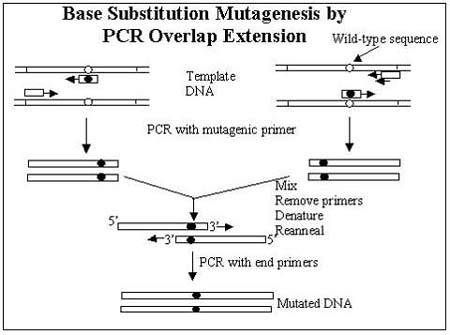
Base substitution mutations can be introduced by PCR overlap extension mutagenesis. Two separate PCRs are performed, each using a perfectly complementary primer at the end of the sequence and a mismatched primer designed to introduce a point mutation at a specific point. This results in two overlapping fragments that contain the base substitution. The fragments are then annealed together in a secondary PCR to amplify the complete mutated gene
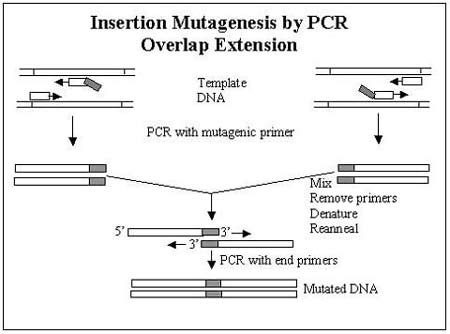
Overlap extension can be used to introduce longer sequences by amplifying the insertion sequence and the flanking sequence, then annealing them together, but this can be time-consuming and difficult.
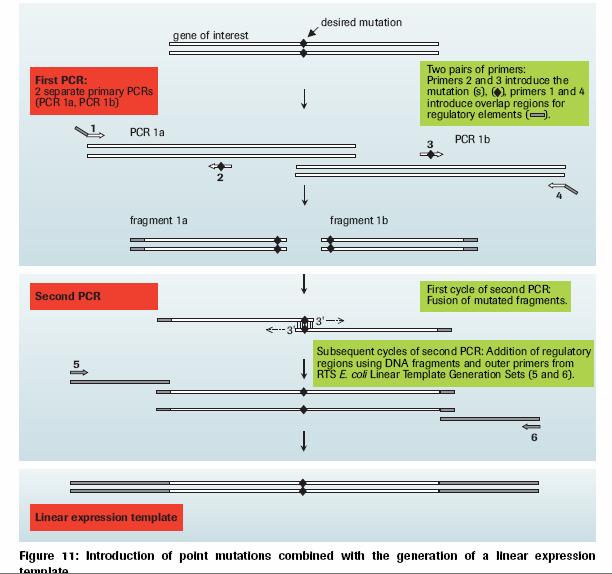
LA-PCR = 'Long and Accurate PCR technology'
This method needs only a single mutagenic primer (+ 3 other primers which are the same for all constructs if used in the same vector) and would be a cheap and reliable method if lots of mutants need to be made.
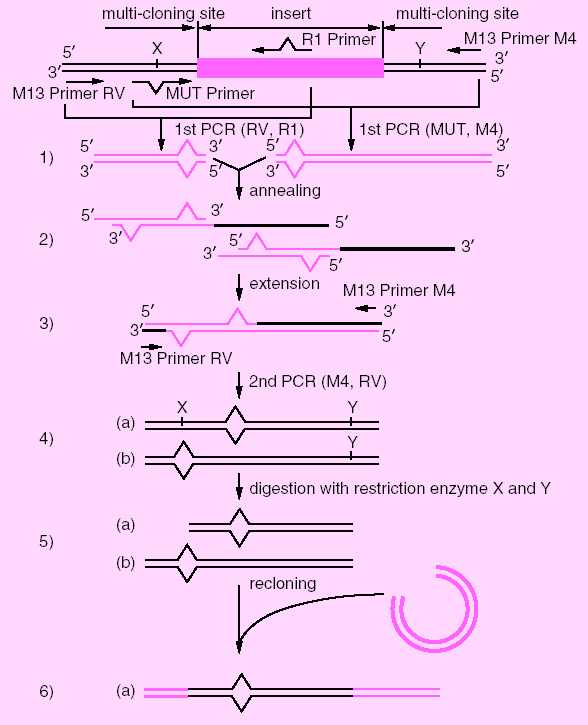
1) Insert target DNA into multicloning site of pUC vectors. Choose one of MUT Primers to destroy a restriction site based on the direction of R1 Primer (primer for introducing a mutation) and the restriction site used for DNA insertion. Perform the first PCR by the combination of R1 Primer and M13 Primer RV (or M13 Primer M4), MUT Primer and M13 Primer M4 (or M13 Primer RV) separately in two tubes.
2) After the elimination of the excess amount of the primers,
amplified products are mixed, heat denatured and annealed.
3) Add TaKaRa LA Taq to complete heterogeneous double strand.
4) Perform the second PCR by using M13 Primer M4 and M13 Primer RV, which will result in two types of the amplified products (a) and (b).
5) Digest the amplified products with two restriction
enzymes, one of which should recognize the site (X) that had been destroyed by
MUT Primer and the other should recognize the appropriate site (Y) within the
multicloning site.
6) Reclone the digested fragment into the vector digested with the same two
restriction enzymes. Only the fragment (a), which contains the mutation
introduced by R1 Primer sequence, will be recloned.
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 9858
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved