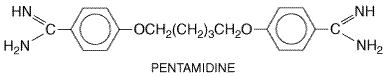| CATEGORII DOCUMENTE |
| Bulgara | Ceha slovaca | Croata | Engleza | Estona | Finlandeza | Franceza |
| Germana | Italiana | Letona | Lituaniana | Maghiara | Olandeza | Poloneza |
| Sarba | Slovena | Spaniola | Suedeza | Turca | Ucraineana |
Drugs Used in the Chemotherapy of Protozoal Infections: Amebiasis, Giardiasis, Trichomoniasis, Trypanosomiasis, Leishmaniasis, and Other Protozoal Infections
Overview
|
Human beings host a wide variety of protozoal parasites that can be transmitted by insect vectors, directly from other mammalian reservoirs, or from one person to another. Because protozoa multiply rapidly in their hosts and effective vaccines are as yet unavailable, chemotherapy has been the only practical way to both treat infected individuals and reduce transmission. The immune system plays a crucial role in protecting against the pathological consequences of protozoal infection. Thus, opportunistic infections with protozoa are prominent in infants, individuals with cancer, transplant recipients, those receiving immunosuppressive drugs or extensive antibiotic therapy, and persons with advanced human immunodeficiency virus (HIV) infection. Treatment of protozoal infections in immunocompromised individuals is especially difficult, and the outcome is often unsatisfactory. Most antiprotozoal drugs have been in use for years despite major advances in bioscience relevant to parasite biology, host defenses, and mechanisms of disease. Satisfactory agents for treating important protozoal infections such as African trypanosomiasis (sleeping sickness) and chronic Chagas' disease still are lacking. Many effective antiprotozoal drugs are toxic at therapeutic doses, a problem exacerbated by increasing drug resistance. Development of drug resistance also poses a serious threat to better-tolerated antiprotozoal agents in current use. This chapter briefly describes important human protozoal infections other than malaria and features the drugs used to treat them. Presented first are amebiasis, giardiasis, and trichomoniasis, three cosmopolitan infections caused by anaerobic protozoa. Descriptions of toxoplasmosis and cryptosporidiosis follow: these infections especially threaten immunocompromised individuals such as those with acquired immunodeficiency syndrome (AIDS). Next are trypanosomiasis and leishmaniasis, two devastating infections caused by different Kinetoplastidae that affect millions of people in tropical regions. Mention is then made of far less-common protozoal infections of human beings such as balantidiasis and babesiosis. The main text deals with the properties and uses of primary drugs for these infectionsi.e., diloxanide furoate, eflornithine, melarsoprol, metronidazole, nifurtimox, pentamidine, sodium stibogluconate, and suramin. Less attention is devoted to secondary and historical drugs used for these infectionsi.e., chloroquine, emetine/dehydroemetine, iodoquinol, quinacrine, and the antiprotozoal antibiotics. The chapter concludes with a brief prospectus about the future of antiprotozoal chemotherapy. |
Introduction to Protozoal Infections of Human Beings
|
Amebiasis Amebiasis affects about 10% of the world's population, causing
invasive disease in about 50 million people and death in about 100,000 of
these annually. Although endemic amebiasis is relatively rare in the general
population of the Drugs used to treat amebiasis can be categorized as luminal,
systemic, or mixed amebicides. Luminal amebicides, exemplified by diloxanide
furoate, iodoquinol, and the nonabsorbed aminoglycoside paromomycin,
are active against only intestinal forms of amoebae. These compounds can be
used successfully by themselves to treat asymptomatic or mild intestinal
forms of amebiasis or after a systemic or mixed amebicide to eradicate the
infection. Systemic amebicides are effective only against invasive forms of
amebiasis. These agents have been employed primarily to treat severe amebic
dysentery (dehydroemetine) or hepatic abscesses (dehydroemetine or chloroquine),
but they are not recommended unless other drugs fail or cause unacceptable
side effects. Mixed amebicides are active against both intestinal and
systemic forms of amebiasis. Metronidazole, a nitroimidazole
derivative, is the prototypical mixed amebicide available in the Giardiasis Giardiasis, caused by the flagellated protozoan Giardia lamblia,
is prevalent worldwide and is also the most commonly reported intestinal
protozoal infection in the Trichomoniasis Trichomoniasis is caused by the flagellated protozoan Trichomonas
vaginalis. This organism inhabits the genitourinary tract of the human
host, where it causes vaginitis in women and, uncommonly, urethritis in men.
Transmission of the infection occurs by sexual contact, and over 200 million
people worldwide become infected each year. In the Toxoplasmosis Toxoplasmosis is a cosmopolitan zoonotic infection caused by the obligate intracellular protozoan Toxoplasma gondii (seeWong and Remington, 1993). Although cats and other feline species are the natural hosts, tissue cysts (bradyzoites) have been recovered from all mammalian species examined. The four most common routes of infection in human beings are (1) ingestion of undercooked meat containing tissue cysts, (2) ingestion of vegetable matter contaminated with soil containing infective oocysts, (3) direct oral contact with feces of cats shedding oocysts, and (4) transplacental fetal infection with tachyzoites from acutely infected mothers. Toxoplasmosis produces clinical symptoms in only about 10% to 20% of
immunocompetent individuals, although close to 70% of adults in the Cryptosporidiosis Coccidian protozoan parasites of the genus Cryptosporidium have been detected in mammals, birds, fish, and reptiles. Recognized as human pathogens in 1976, these enteric organisms can cause severe bouts of watery diarrhea in both domestic animals and human beings (seeGriffiths, 1998). Infectious oocysts in feces may be spread either by direct human-to-human contact or by contaminated water supplies, the latter an established route of epidemic infection. Groups at risk include travelers, children in day-care facilities, male homosexuals, animal handlers, veterinarians, and other health care personnel. Immunocompromised individuals are especially vulnerable. After ingestion, the mature oocyte is digested, releasing sporozoites that invade host epithelial cells, penetrating the cell membrane but not actually entering the cytoplasm. In most individuals, infection is self-limited. However, in AIDS patients and other immunocompromised individuals, the severity of voluminous, secretory diarrhea usually requires hospitalization and supportive therapy to prevent severe electrolyte imbalance and dehydration. Combined therapy with paromomycin and azithromycin may be beneficial in some AIDS patients suffering from chronic cryptosporidiosis (Smith et al., 1998). However, there is currently no known effective drug for treatment of cryptosporidiosis. Trypanosomiasis African trypanosomiasis or 'sleeping sickness' is caused by
subspecies of the hemoflagellate Trypanosoma brucei that are
transmitted by bloodsucking tsetse flies of the genus Glossinia.
Largely restricted to central American trypanosomiasis or Chagas' disease, a zoonotic infection caused by Trypanosoma
cruzi, affects about 24 million people from southern California to
Argentina and Chile (seeTanowitz et al., 1992; Kirchhoff, 1996),
where the chronic form of the disease in adults is a major cause of
cardiomyopathy, megaesophagus, megacolon, and death. Bloodsucking triatomid
bugs infesting poor rural dwellings most commonly transmit this infection to
young children; transplacental transmission also may occur in endemic areas. Acute
infection is evidenced by a raised tender skin nodule (chagoma) at the
site of inoculation; other signs may be absent or range from fever, adenitis,
skin rash, and hepatosplenomegaly to, albeit rarely, acute myocarditis and
death. Invading metacyclic trypomastigotes penetrate host cells,
especially macrophages, where they proliferate as amastigotes. The
latter then differentiate into trypomastigotes that enter the bloodstream.
Circulating trypomastigotes do not multiply until they invade other cells or are
ingested by an insect vector during a blood meal. After recovery from the
acute infection within a few weeks to months, individuals usually remain
asymptomatic for years despite sporadic parasitemia. During this period their
blood can transmit the parasites to transfusion recipients and accidentally
to laboratory workers. An increasing fraction of adults develop overt chronic
disease of the heart and gastrointestinal tract as they age. Progressive
destruction of myocardial cells and neurons of the myenteric plexus results
from the special tropism of T. cruzi for muscle cells. Whether or not
an undefined autoimmune response also contributes to the pathogenesis of
Chagas' disease is controversial, especially since recent studies with
improved techniques indicate the presence of T. cruzi at sites of
cardiac lesions (Urbina, 1999). However, immunological defenses, especially
cell-mediated immunity, do play a role in modulating the course of disease.
Two nitroheterocyclic drugs, nifurtimox, which is available from the
Centers for Disease Control and Prevention (CDC), and benznidazole,
which is not, are used to treat this infection. Both agents suppress
parasitemia and may even cure the acute phase of Chagas' disease, but they
are far less effective against the chronic infection (Kirchhoff, 1996). Both
drugs are toxic and must be taken for long periods. Field isolates vary with
respect to their susceptibility to nifurtimox and benznidazole. Moreover,
resistance to both compounds can be induced in the laboratory. While both
drugs can generate intracellular free radicals, their mechanisms of action
and resistance are not well understood. Drug development for Chagas' disease
has lagged due to lack of economic incentives, even though T. cruzi
offers a variety of potential therapeutic targets (seeUrbina, 1999).
Indeed, alternative measures such as improved vector control and housing
accommodations have substantially reduced transmission of Chagas' disease in Leishmaniasis Leishmaniasis is a complex, vector-borne zoo-nosis caused by about 20
different species of obligate intramacrophage protozoa of the genus Leishmania.
Small mammals and canines generally serve as reservoirs for these pathogens,
which can be transmitted to human beings by the bites of some 30 different
species of female phlebotomine sandflies. Various forms of leishmaniasis
affect people in southern The classification, clinical features, course, and chemotherapy of the
various human leishmaniasis syndromes have been reviewed recently, in
addition to the biochemistry and immunology of the parasite and host germane
to chemotherapy (seeHerwaldt, 1999b). Cutaneous forms of leishmaniasis
generally are self-limiting, whereas the mucocutaneous, diffuse cutaneous,
and visceral forms are not. Initial parenteral treatment with pentavalent
antimonials, according to regimens based on collective empirical
experience, appears safe and effective in most cases, but prolonged therapy
is required, and resistance to these agents is increasing. Amphotericin B
and pentamidine, formerly judged as secondary drugs because of
unacceptable toxicity at therapeutic doses, are now undergoing reevaluation
because of improved formulations and dosage schedules. For example, in 1997
the FDA approved a lipid formulation of amphotericin B (AMBISOME; seeChapter 49:
Antimicrobial Agents: Antifungal Agents) for therapy of visceral
leishmaniasis. Now considered a first-line drug for this indication, lipid
formulations of amphotericin B can be especially useful for patients who fail
antimonial therapy or who cannot tolerate long courses of parenteral therapy
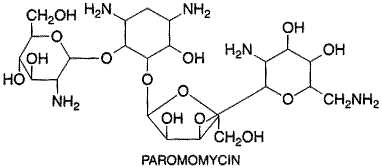
(seeMeyerhoff, 1999; Herwaldt, 1999b). The aminoglycoside paromomycin
has been used parenterally as monotherapy for visceral leishmaniasis in Other Protozoal Infections Just a few of the many less-common protozoal infections of human beings are highlighted here. The reader is referred to the 14th edition of Harrison's Principles of Internal Medicine for more details and to Rosenblatt (1999) for specific therapeutic regimens. Babesiosis, caused by either Babesia microcoti or B. divergens, is a tick-borne zoonosis that superficially resembles malaria in that the parasites invade erythrocytes, producing a febrile illness, hemolysis, and hemoglobinuria. This infection usually is mild and self-limiting but can be severe or even fatal in asplenic or severely immunocompromised individuals. The macrolide antibiotic azithromycin has been used successfully along with either quinine or atovaquone (seeChapter 40: Drugs Used in the Chemotherapy of Protozoal Infections: Malaria) to treat babesiosis in experimental animals and in some patients; chloroquine is not effective. Gastrointestinal infections caused by a variety of pathogenic protozoa can be especially severe in immunocompromised patients such as those with AIDS. Whereas chemotherapy of cryptosporidiosis has proven to be difficult in this population, it has produced better responses in two other coccidian infections of human beings, i.e., isosporiasis and cyclosporiasis. Thus, trimethoprim-sulfamethoxazole has been found successful for controlling diarrhea due to Isospora belli in AIDS patients, even though relapses may occur and long-term maintenance therapy may be required. Pyrimethamine has been used to treat those patients who cannot tolerate sulfonamides. Trimethoprim-sulfamethoxazole also is effective against Cyclospora cayatensis, an organism that can produce prolonged or relapsing diarrhea in travelers or AIDS patients. Microsporidiosis, a transient infection in travelers to the tropics but a major cause of diarrhea in immunocompromised individuals, can be caused by several different genera of microsporidian parasites that respond in varying degrees to the benzimidazole anthelmintic albendazole (seeChapter 42: Drugs Used in the Chemotherapy of Helminthiasis), given alone or with other agents such as furazolidone. Balantidiasis, caused by the ciliated protozoan Balantidium coli, is an infection of the large intestine that may be confused with amebiasis. Unlike amebiasis, however, this infection usually responds to tetracycline therapy. |
Chloroquine
|
The pharmacology and toxicology of chloroquine are presented in Chapter 40: Drugs Used in the Chemotherapy of Protozoal Infections: Malaria. Only those features of the drug pertinent to its use in amebiasis are described here. The unique therapeutic value of chloroquine for extraintestinal amebiasis in human beings relates to its direct toxic action against trophozoites of E. histolytica together with the fact that it is highly concentrated in liver. Chloroquine is used as a systemic amebicide to treat hepatic amebiasis only when treatment with metronidazole is unsuccessful or contraindicated. The clinical response to chloroquine in patients with hepatic amebiasis is usually prompt, and there is no evidence that amoebae develop resistance to this agent. The drug is far less effective in intestinal amebiasis, because it is almost completely absorbed from the small bowel and attains only low concentrations in the intestinal wall. Colonic infection with E. histolytica is always the source of extraintestinal amebiasis, so a drug effective in intestinal amebiasis is given routinely to all patients receiving chloroquine for hepatic amebiasis; such therapy reduces the relapse rate. The conventional course of treatment with chloroquine phosphate for extraintestinal amebiasis in adults is 1 g daily for 2 days, followed by 500 mg daily for at least 2 to 3 weeks. Because of the low toxicity of this drug, this dose can be increased or the schedule can be repeated if necessary. |
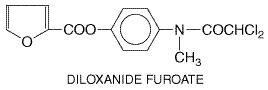
Diloxanide Furoate
|
History Diloxanide
is a dichloroacetamide derivative that was identified as a result of the
examination of a series of substituted acetanilides for amebicidal activity.
Of the many derivatives of diloxanide prepared, the furoate ester proved to
be appreciably more active than the parent compound in experimentally
infected rats (Main et al., 1960). The results of clinical trials
showed it to be effective in cases of acute intestinal amebiasis. Diloxanide
furoate (FURAMIDE),
which is not available in the
Pharmacological Effects Diloxanide is directly amebicidal when tested in vitro. The
furoate ester is active at 0.01 to 0.1 Absorption, Fate, and Excretion After oral ingestion, the ester is largely hydrolyzed in the lumen or mucosa of the intestine to diloxanide and furoic acid; only diloxanide appears in the systemic circulation. In experimental animals, 60% to 90% of an oral dose is excreted in the urine within 48 hours, chiefly as the glucuronide. More than half of this appears within 6 hours. Excretion in the feces accounts for 4% to 9% of the dose. The blood concentration of diloxanide peaks within 1 hour but falls to a fraction of this level within 6 hours. Therapeutic Uses Given alone, diloxanide furoate is effective for treatment of asymptomatic passers of amebic cysts (Krogstad et al., 1978). Other drugs effective in asymptomatic amebiasis are iodoquinol and paromomycin (Anonymous, 1998). Diloxanide is ineffective when administered alone in the treatment of extraintestinal amebiasis, and its efficacy when used alone in the treatment of acute amebiasis with frank dysentery is controversial. Although good results have been reported in some areas, other trials have been less successful (Suchak et al., 1962). In trials carried out primarily in asymptomatic subjects passing trophozoites or cysts, or in patients with nondysenteric, symptomatic intestinal amebiasis, treatment with diloxanide furoate resulted in a high percentage of cures (Wolfe, 1973). Diloxanide furoate is used along with or after an appropriate systemic or mixed amebicide to effect a cure of invasive and extraintestinal amebiasis. Diloxanide furoate is given orally. The recommended dose for adults is 500 mg three times daily for 10 days. If necessary, treatment can be extended to 20 days. Children should be given 20 mg/kg per day in three divided doses for 10 days. Toxicity and Side Effects Diloxanide furoate generally is well tolerated and side effects are mild. Flatulence is most commonly reported; nausea, vomiting, diarrhea, pruritus, and urticaria occur occasionally (seeWolfe, 1973) |
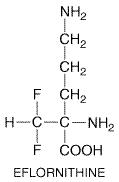
Eflornithine
|
History Eflornithine
Antitrypanosomal Effects The effects of eflornithine have been evaluated both on drug-susceptible and drug-resistant T. brucei in vitro and on infections with these parasites in rodent models. This cytostatic agent has multiple biochemical effects on trypanosomes. Not only is polyamine and trypanothione biosynthesis reduced and methionine metabolism altered, but macromolecular biosynthesis is generally depressed and cell division ceases. Trypanosomes exposed to eflornithine change from long, slender, quickly dividing bloodstream forms that avoid host defenses by rapidly synthesizing variable cell-surface glycoproteins to short, nonreplicating forms that fail to synthesize these molecules and are rapidly cleared from the circulation (Wang, 1997). Molecular mechanisms of eflornithine action and resistance in African trypanosomes, though complex, are becoming better understood, as indeed are the reasons for the drug's greater effectiveness against T.b. gambiense than against T.b. rhodesiense (reviewed by Wang, 1997). Eflornithine irreversibly inhibits both mammalian and trypanosomal ornithine decarboxylases, thereby preventing the synthesis of putrescine, a precursor of polyamines needed for cell division. However, both the mammalian host and T.b. rhodesiense replace the inhibited enzyme far more rapidly than T.b. gambiense, and T.b. rhodesiense has higher levels of ornithine decarboxylase activity than doesT.b. gambiense. Both findings are consistent with the selective trypanostatic action of eflornithine in T.b. gambiense (seeWang, 1997; Iten et al., 1998). The slender bloodstream forms of human trypanosomes must synthesize polyamines de novo, because human blood contains only very low levels of these essential compounds. Mutant bloodstream trypanosomes lacking ornithine decarboxylase or wild-type trypanosomes treated with eflornithine convert into nonreplicating, noninfectious, stationary parasites that are rapidly cleared from the blood (Li et al., 1998). Absorption, Fate, and Excretion Eflornithine is given by either the intravenous or the oral route; its bioavailability after oral administration is about 54%. Peak plasma levels are achieved about 4 hours after an oral dose, and the elimination half-life averages about 200 minutes. The drug does not bind to plasma proteins, but it is well distributed and penetrates into the cerebrospinal fluid. The last property is especially important in late-stage African trypanosomiasis, where cerebrospinal fluid/plasma ratios exceeding 0.9 have been reported. Over 80% of eflornithine is cleared by the kidney, largely in unchanged form. There is some evidence that eflornithine displays dose-dependent pharmacokinetics at the highest doses used clinically (seeAbeloff et al., 1984; Ppin and Milord, 1994). Therapeutic Uses Experience with the use of eflorni-thine for the treatment of West African trypanosomiasis due to T.b. gambiense has been well summarized by van Nieuwenhove (1992) and by Ppin and Milord (1994). Most patients reported had advanced disease with CNS complications, and many had received arsenicals prior to treatment with eflornithine. The preferred regimen for adult patients was found to be 100 mg/kg given intravenously every 6 hours for 14 days. Virtually all patients improved on this regimen unless they were extremely ill; the probable cure rate exceeded 60%. Whether or not a shorter course of therapy would be equally effective is not known. Children younger than 12 years old required higher doses of eflornithine, probably because they clear the drug more rapidly than do adults and because the drug does not reach the CNS as well. To avoid early convulsions, which could be more frequent in children receiving higher doses, a regimen using the current intravenous dosage (400 mg/kg per day) in the first few days, followed by an increase for the second part of therapy, has been proposed (Milord et al., 1993). Equal doses of eflornithine were less effective when given by the oral route, probably because of limited bio-availability. The problem cannot be overcome simply by increasing the oral dose, because of ensuing osmotic diarrhea. However, the oral route can be used when intravenous therapy cannot be instituted. A relapse rate of about 15% was estimated for patients taking 100 mg orally every 6 hours for 21 to 45 days. Relapses occurred in only about 5% of patients receiving the optimal 14-day intravenous regimen. Eflornithine has proven to be less successful for treating AIDS patients with West African trypanosomiasis, presumably because host defenses play a critical role in clearing drug-treated T.b. gambiense from the bloodstream. Even high doses of eflornithine failed to improve East African trypanosomiasis due to T.b. rhodesiense, consistent with the relatively short half-life and high activity of ornithine decarboxylase in these parasites. Eflornithine alone appears to be rather ineffective for therapy of human leishmaniasis and P. carinii pneumonia, even though experimental evidence indicates that this compound can deplete polyamines in Leishmania spp. and P. carinii. Toxicity and Side Effects Eflornithine causes a wide array of adverse effects in treated patients (seevan Nieuwenhove, 1992; Ppin and Milord, 1994). Anemia (48%), diarrhea (39%), and leukopenia (27%) are the most common complications in patients receiving intravenous medication. Diarrhea is both dose-related and doselimiting, especially after oral administration of the drug. Convulsions occur early in about 7% of treated patients, but they do not appear to recur despite continuation of therapy. Other complicationssuch as thrombocytopenia, alopecia, vomiting, abdominal pain, dizziness, fever, anorexia, and headacheoccur in less than 10% of treated patients. Most of the above side effects are reversed by withdrawal of the drug. Patients are not routinely tested for hearing loss, but this reversible complication can occur after prolonged therapy with low oral doses (Pasic et al., 1997). Eflornithine interferes with normal embryonic development in experimental animals. Therapeutic doses of eflornithine are large and require coadministration of substantial volumes of intravenous fluid. This can pose practical limitations in remote settings and cause fluid overload in susceptible patients. In any event, the risks may outweigh the benefits if eflornithine therapy is continued beyond 21 days. |
Emetine and Dehydroemetine
|
The use of emetine, an alkaloid derived from ipecac
(' |
8-Hydroxyquinolines
|
A number of halogenated 8-hydroxyquinolines
have been synthesized and used clinically as luminal amebicides, especially to
treat asymptomatic cyst passers. Such direct-acting amebicidal agents also
have been used together with metronidazole to treat intestinal forms of
amebiasis. Iodoquinol (diiodohydroxyquin) and clioquinol
(iodochlorhydroxyquin; available in the |
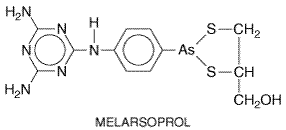
Melarsoprol
|
History In 1949, Friedheim demonstrated that melarsoprol, the dimercaptopropanol derivative of melarsen oxide, was effective in the treatment of advanced cases of trypanosomiasis. It was considerably safer than other trypanocides available at the time and has remained a first-line drug in the treatment of late (CNS) stages of both West and East African trypanosomiasis. Chemistry and Preparation Melarsoprol has the following chemical structure:
Melarsoprol (Mel B;ARSOBAL), consisting of two stereoisomers in a 3:1
ratio (Ericsson et al., 1997), is insoluble in water and is supplied
as a 3.6% (w/v) solution in propylene glycol for intravenous administration.
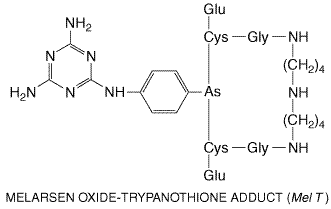
It is available in the Antiprotozoal Effects It is the trivalent arsenoxide form of an organic arsenical that accounts for both its rapid lethal effect on African trypanosomes and its toxicity to the host (seeAlbert, 1979). Arsenoxides react avidly and reversibly with vicinal sulfhydryl groups, including those of proteins, and thereby inactivate a great number and variety of enzymes. The same nonspecific mechanism by which melarsoprol is lethal to parasites is probably responsible for its toxicity to host tissues. However, susceptible African trypanosomes actively concentrate melarsoprol via an unusual purine transporter (Carter and Fairlamb, 1993; Barrett and Fairlamb, 1999). The basis for the trypanocidal action of melarsoprol is not understood, probably due to its high reactivity with many biomolecules. For example, melarsoprol is a potent inhibitor of pyruvate kinase (Flynn and Bowman, 1969), and disruption of energy metabolism by inhibition of glycolysis was long thought to explain its trypanocidal activity. Other evidence suggests, however, that this is not a primary effect (Van Schaftigen et al., 1987; Eisenthal and Cornish-Bowden, 1998). In a series of studies, Fairlamb and coworkers found that melarsoprol reacts with an unusual trypanosomal dithiol, trypanothione, a spermidine-glutathione adduct. Trypanothione substitutes for glutathione in trypanosomes and other Kinetoplastida to maintain an intracellular reducing environment. Binding of melarsoprol to trypanothione results in formation of melarsen oxide-trypanothione adduct (Mel T), a compound that is a potent competitive inhibitor of trypanothione reductase, the enzyme responsible for maintaining trypanothione in its reduced form. However, critical evidence directly linking melarsoprol's action on the trypanothione system to parasite death is still lacking (seeBarrett and Fairlamb, 1999). At concentrations of 0.5 to 10
Resistance to melarsoprol can result from altered drug uptake via an unusual purine transporter (Carter and Fairlamb, 1993). Moreover, cross-resistance between arsenicals and diamidines (pentamidine) in cloned lines of T. brucei suggests that these drugs are concentrated by the same transport system (seeBarrett and Fairlamb, 1999). Absorption, Fate, and Excretion Melarsoprol is always administered intravenously. A small but therapeutically significant amount of the drug enters the cerebrospinal fluid and has a lethal effect on trypanosomes infecting the CNS. The compound is excreted rapidly, with 70% to 80% of the arsenic appearing in the feces (seePpin and Milord, 1994). Therapeutic Uses Melarsoprol is the only effective drug available for treatment of the late meningoencephalitic stage of both West African (Gambian) and East African (Rhodesian) trypanosomiasis. Also effective in the early hemolymphatic stage of these infections, melarsoprol is reserved for therapy of late-stage infections because of its toxicity. Treatment of East African trypanosomiasis with melarsoprol is initiated soon after the diagnosis is made, because CNS involvement occurs early in this aggressive infection. Melarsoprol is not used for prophylaxis of trypanosomiasis because of its toxicity and rapid elimination. The pattern of resistance to melarsoprol therapy differs between the two subspecies of T. brucei. Patients infected with T.b. rhodesiense who relapse after a course of melarsoprol usually respond to a second course of the drug. In contrast, patients infected with T.b. gambiense who are not cured with melarsoprol rarely benefit from repeated treatment with this drug. Such patients often respond well to eflornithine, which is ineffective against T.b. rhodesiense (Ppin and Milord, 1994). Treatment schedules with melarsoprol were derived empirically more than 40 years ago, and they have not changed appreciably since (seePpin and Milord, 1994). Melarsoprol is administered by slow intravenous injection; care must be taken to avoid leakage into the surrounding tissues, because the drug is intensely irritating. Therapeutic regimens are complex and difficult to individualize because melarsoprol has such a narrow therapeutic window (i.e., low doses risk therapeutic failure, whereas higher doses can cause reactive encephalopathy in 4% to 10% of treated patients). For example, a regimen recommended for patients with advanced meningoencephalitis and those who are febrile consists of pretreatment with suramin (5 mg/kg, 10 mg/kg, and 20 mg/kg intravenously on days 1, 3, and 5) followed by four series of escalating intravenous doses of melarsoprol (0.36 mg/kg, 0.72 mg/kg, and 1.1 mg/kg on days 7, 8, and 9; 1.8 mg/kg on days 16, 17, and 18; 2.2 mg/kg on day 25; 2.9 mg/kg on day 26; 3.6 mg/kg on day 27; and 3.6 mg/kg on days 34, 35, and 36, to a total maximum daily dose of 180 mg). A shorter course of therapy with higher doses has been used for patients in generally good condition. This consists of suramin pretreatment (5 mg/kg and 10 mg/kg intravenously on days 1 and 3) followed by three series of melarsoprol doses given intravenously (1.4 mg/kg, 1.8 mg/kg, and 2.2 mg/kg on days 5, 6, and 7; 2.5 mg/kg, 2.9 mg/kg, and 3.3 mg/kg on days 14, 15, and 16; and 3.6 mg/kg on days 23, 24, and 25, to a maximum daily dose of 180 mg). Lesser doses should be given to children and debilitated patients. Unless contraindicated, pretreatment with glucocorticoids should be initiated 48 hours before suramin to decrease the incidence of reactive encephalopathy. Although 80% to 90% of patients have been cured by them, such regimens are complex, cumbersome, and difficult to administer. Moreover, based on better pharmacokinetic data, lower doses given over shorter periods may prove to be just as effective. As stated above, those with West African trypanosomiasis who relapse should be treated with eflornithine, whereas those with East African trypanosomiasis often respond favorably to a second course of melarsoprol (seePpin and Milord, 1994). Toxicity and Side Effects Toxicity is common during treatment with melarsoprol (seePpin and Milord, 1994). A febrile reaction often occurs soon after drug injection, especially if parasitemia is high. The most serious complications involve the nervous system. A reactive encephalopathy occurs in about 6% of patients, usually between the first two courses of therapy. This is more common in East African than in West African sleeping sickness and is more likely to develop in patients whose cerebrospinal fluid contains many cells and trypanosomes (Ppin et al., 1995). Manifestations include convulsions associated with acute cerebral edema, rapidly progressive coma, and acute, nonlethal mental disturbances without neurological signs. The reaction often is fatal and may occur in the early hemolymphatic stages as well as in the later CNS stages of the illness. Its cause is unknown, but it may represent an immune reaction elicited by the rapid release of trypanosomal antigens from dying parasites rather than by a direct, toxic effect of the drug. Concurrent administration of prednisolone reduces the frequency of reactive encephalopathy and also can be used to control hypersensitivity reactions that occur most often during the second or subsequent courses of melarsoprol therapy. Peripheral neuropathy, noted in about 10% of patients receiving melarsoprol, probably is due to a direct, toxic effect of the drug. Hypertension and myocardial damage are not uncommon, although shock is rare. Albuminuria occurs frequently, and occasionally the appearance of numerous casts in the urine or evidence of hepatic disturbances may necessitate modification of treatment. Vomiting and abdominal colic also are common, but their incidence can be reduced by injecting melarsoprol slowly into the supine, fasting patient. The patient should remain in bed and not eat for several hours after the injection is given. Precautions and Contraindications Melarsoprol should be given only to patients under hospital supervision so that the dosage regimen may be modified if necessary. It is most important that the initial dosage be based on clinical assessment of the general condition of the patient rather than on body weight. Initiation of therapy during a febrile episode has been associated with an increased incidence of reactive encephalopathy. Administration of melarsoprol to leprous patients may precipitate erythema nodosum. The use of the drug is contraindicated during epidemics of influenza. Severe hemolytic reactions have been reported in patients with deficiency of glucose-6-phosphate dehydrogenase. Pregnancy is not a contraindication for treatment with melarsoprol. |
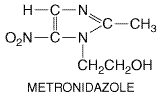
Metronidazole
|
History The isolation of the antibiotic azomycin (2-nitro-imidazole)
from a streptomycete by Maeda and collaborators in 1953 and the demonstration
of its trichomonacidal properties by Horie (1956) led to the chemical
synthesis and biological testing of many nitroimidazoles. One compound, 1-(
Antiparasitic and Antimicrobial Effects Metronidazole and related nitroimidazoles are active in vitro
against a wide variety of anaerobic protozoal parasites and anaerobic
bacteria (seeFreeman et al., 1997). The compound is directly
trichomonacidal. Sensitive isolates of T. vaginalis are killed by
<0.05 Metronidazole manifests antibacterial activity against all anaerobic cocci and both anaerobic gram-negative bacilli, including Bacteroides species, and anaerobic sporeforming gram-positive bacilli. Nonsporulating grampositive bacilli often are resistant, as are aerobic and facultatively anaerobic bacteria. Metronidazole is clinically effective in trichomoniasis, amebiasis, and giardiasis, as well as in a variety of infections caused by obligate anaerobic bacteria, including Bacteroides, Clostridium, and Helicobacter species. Metronidazole may facilitate extraction of adult guinea worms in dracunculiasis, even though it has no direct effect on the parasite (seeChapter 42: Drugs Used in the Chemotherapy of Helminthiasis). Mechanism of Action and Resistance Metronidazole is a prodrug; it requires reductive activation of the nitro group by susceptible organisms. Its selective toxicity towards anaerobic and microaerophilic pathogens such as the amitochondriate protozoa, T. vaginalis, E. histolytica, and G. lamblia, and various anaerobic bacteria derives from their energy metabolism, which differs from that of aerobic cells (seeLand and Johnson, 1997; Samuelson, 1999; Upcroft and Upcroft, 1999). These organisms, unlike their aerobic counterparts, contain electron transport components such as ferredoxins, small Fe-S proteins that have a sufficiently negative redox potential to donate electrons to metronidazole. The single electron transfer forms a highly reactive nitro radical anion that kills susceptible organisms by radical-mediated mechanisms that target DNA and possibly other vital biomolecules. Metronidazole is catalytically recycled; loss of the active metabolite's electron regenerates the parent compound. Increasing levels of O2 inhibit metronidazole-induced cytotoxicity, because O2 competes with metronidazole for electrons generated by energy metabolism. Thus, O2 can both decrease reductive activation of metronidazole and increase recycling of the activated drug. Anaerobic or microaerophilic organisms susceptible to metronidazole derive energy from the oxidative fermentation of ketoacids such as pyruvate. Pyruvate decarboxylation, catalyzed by pyruvate:ferredoxin oxidoreductase, produces electrons that reduce ferredoxin, which catalytically donates its electrons to biological electron acceptors or to metronidazole. Clinical resistance to metronidazole is well documented for T. vaginalis, G. lamblia, and a variety of anaerobic bacteria but has yet to be shown for E. histolytica. Resistance to 5-nitroimidazole drugs in vitro has been studied most extensively with trichomonads, whereas data are limited for both Giardia and amoebae (seeLand and Johnson, 1997; Samuelson, 1999; Kulda, 1999; Upcroft and Upcroft, 1999; Wassmann et al., 1999). Resistant strains of T. vaginalis derived from nonresponsive patients have shown two major types of abnormalities when tested under aerobic conditions. The first correlates with impaired oxygen-scavenging capabilities, leading to higher local O2 concentrations, decreased activation of metronidazole, and futile recycling of the activated drug (see above and Yarlett et al., 1986). The second type is associated with lowered levels for pyruvate:ferredoxin oxidoreductase and ferredoxin, the latter due to reduced transcription of the ferredoxin gene (Quon et al., 1992). That pyruvate:ferredoxin oxidoreductase and ferredoxin are not completely absent may explain why infections with such strains usually respond to higher doses of metronidazole or more prolonged therapy (Johnson, 1993). Whether or not other mechanisms of metronidazole resistance induced by drug exposure in culture actually operate in vivo for trichomonads and amoebae is not known (seeBrown et al., 1999; Wassmann et al., 1999). Absorption, Fate, and Excretion The pharmacokinetic properties of metronidazole and its two major
metabolites have been investigated intensively (seeLamp et al.,
1999). Preparations of metronidazole are available for oral, intravenous,
intravaginal, and topical administration. The drug usually is completely and
promptly absorbed after oral intake, reaching concentrations in plasma of 8
to 13 After an oral dose, over 75% of labeled metronidazole is eliminated in the urine, largely as metabolites; only about 10% is recovered as unchanged drug. The liver is the main site of metabolism, and this accounts for over 50% of the systemic clearance of metronidazole. The two principal metabolites result from oxidation of side chains, a hydroxy derivative and an acid. The hydroxy metabolite has a longer half-life (about 12 hours) and nearly 50% of the antitrichomonal activity of metronidazole. Formation of glucuronides also is observed. Small quantities of reduced metabolites, including ring-cleavage products, are formed by the gut flora. The urine of some patients may be reddish-brown owing to the presence of unidentified pigments derived from the drug. Oxidative metabolism of metronidazole is induced by phenobarbital, prednisone, rifampin, and possibly ethanol. Cimetidine appears to inhibit hepatic metabolism of the drug. Therapeutic Uses The uses of metronidazole for antiprotozoal therapy have been extensively reviewed (seeFreeman et al., 1997; Johnson, 1993; Ravdin, 1995; Zaat et al., 1997). Metronidazole cures genital infections with T. vaginalis in both females and males in more than 90% of cases. The preferred treatment regimen is 2 g of metronidazole as a single oral dose for both males and females. For patients who cannot tolerate a single 2-g dose, an alternative regimen is a 250-mg dose given three times daily or a 375-mg dose given twice daily for 7 days. When repeated courses or higher doses of the drug are required for uncured or recurrent infections, it is recommended that intervals of 4 to 6 weeks elapse between courses. In such cases, leukocyte counts should be carried out before, during, and after each course of treatment. Lack of satisfactory response may be due to bacterial vaginosis, chronic infection of the cervical glands or of Skene's and Bartholin's glands. Reinfection by an infected partner also may cause an unsatisfactory response. Although once rare, treatment failures due to the presence of metronidazole-resistant strains of T. vaginalis are becoming increasingly common. Most of these cases can be treated successfully by giving a second 2-g dose to both patient and sexual partner. In addition to oral therapy, the use of topical gel containing 0.75%metronidazole or a 500 to 1000 mg vaginal suppository will increase the local concentration of drug and may be beneficial in refractory cases (Heine and McGregor, 1993). Metronidazole is an effective amebicide and has become the agent of choice for the treatment of all symptomatic forms of amebiasis, including acute gastrointestinal infection and liver abscess. The recommended dose is 500 to 750 mg of metronidazole taken orally three times daily for 10 days. The daily dose for children is 35 to 50 mg/kg given in three divided doses for 10 days. E. histolytica persist in most patients who recover from acute amebiasis after metronidazole therapy, so it is recommended that all such individuals be treated with a luminal amebicide such as diloxanide furoate. Clinical resistance of E. histolytica to metronidazole has not been confirmed, despite extensive clinical use of this drug. Mass treatment with a large dose of metronidazole once monthly for a few months and then in alternate months has resulted in a marked decrease in the incidence of amebic dysentery in relatively isolated communities with a high degree of endemicity. Although effective for the therapy of giardiasis, metronidazole has
yet to be approved for treatment of this infection in the Metronidazole is a relatively inexpensive, highly versatile drug with clinical efficacy against a broad spectrum of anaerobic bacteria (seeFreeman et al., 1997). The compound is used for treatment of serious infections due to susceptible anaerobic bacteria, including Bacteroides, Clostridium, Fusobacterium, Peptococcus, Peptostreptococcus, Eubacterium, and Helicobacter. The drug also can be given along with other antimicrobial agents to treated mixed infections with aerobic and anaerobic bacteria. Due to excellent tissue penetration, metronidazole can achieve clinically effective levels at sites such as bones, joints, and the brain. Intraabdominal, gynecologic, dermal, and CNS infections with susceptible anaerobes all have responded to this drug, as have bacterial septicemia and endocarditis. Metronidazole can be given intravenously when oral administration is not indicated. A typical intravenous regimen for severe anaerobic infections is a loading dose of 15 mg/kg followed 6 hours later by a maintenance dose of 7.5 mg/kg every 6 hours, usually for 7 to 10 days. Together with other antibiotics, metronidazole has shown efficacy for prophylaxis of postsurgical mixed bacterial infections (Song and Glenny, 1998) and for treatment of gastric infections with H. pylori when taken in various regimens that include the proton pump inhibitors (Hopkins and Morris, 1994; Harris, 1998; Megraud and Doermann, 1998; seeChapter 37: Agents Used for Control of Gastric Acidity and Treatment of Peptic Ulcers and Gastroesophageal Reflux Disease). Because of its low cost, the drug has also been used instead of vancomycin to treat pseudomembranous colitis. Metronidazole and other nitroimidazoles can sensitize hypoxic tumor cells to the effects of ionizing radiation, but these drugs are not used clinically for this purpose. Toxicity, Contraindications, and Drug Interactions The toxicity of metronidazole has been reviewed (seeRoe, 1977; Lau et al., 1992). Side effects only rarely are severe enough to discontinue therapy. The most common are headache, nausea, dry mouth, and a metallic taste. Vomiting, diarrhea, and abdominal distress occasionally are experienced. Furry tongue, glossitis, and stomatitis occurring during therapy usually are associated with an exacerbation of moniliasis. Dizziness, vertigo, and, very rarely, encephalopathy, convulsions, incoordination, and ataxia are neurotoxic effects that warrant discontinuation of metronidazole. The drug also should be withdrawn if numbness or paresthesias of the extremities occur. Reversal of serious sensory neuropathies may be slow or incomplete. Urticaria, flushing, and pruritus are indicative of drug sensitivity that can require withdrawal of metronidazole. Dysuria, cystitis, and a sense of pelvic pressure also have been reported. Metronidazole has a well-documented disulfiram-like effect, such that some patients experience abdominal distress, vomiting, flushing, or headache if they drink alcoholic beverages during or within 3 days after therapy with this drug. Patients should be cautioned to avoid consuming alcohol during metronidazole treatment, even though the risk of a severe reaction is low. By the same token, metronidazole and disulfiram should not be taken together, because confusional and psychotic states may occur. Although related chemicals have caused blood dyscrasias, only a temporary neutropenia, reversible after discontinuation of therapy, occurs with metronidazole. Metronidazole should be used with caution in patients with active disease of the CNS because of its potential neurotoxicity. The drug also may precipitate CNS signs of lithium toxicity in patients receiving high doses of lithium. Plasma levels of metronidazole can be elevated by drugs such as cimetidine that inhibit hepatic microsomal metabolism. Moreover, metronidazole can prolong the prothrombin time of patients receiving therapy with coumadin anticoagulants. The dosage of metronidazole should be reduced in patients with severe hepatic disease. Given in high doses for prolonged periods, metronidazole is carcinogenic in rodents; it also is mutagenic in bacteria (seeLau et al., 1992). Mutagenic activity is associated with metronidazole and several of its metabolites found in the urine of patients treated with therapeutic doses of the drug. However, there is no evidence that therapeutic doses of metronidazole pose any significant increased risk of cancer to human patients. There is conflicting evidence about the teratogenicity of metronidazole in animals. While metronidazole has been taken during all stages of pregnancy with no apparent adverse effects, its use during the first trimester is not advised. |
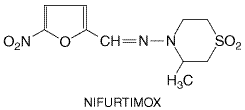
Nifurtimox
|
History Nitrofurans were known to be effective in experimental infections with
American trypanosomiasis caused by T. cruzi, so numerous congeners
were tested for their chemotherapeutic potential. Of these, one drug, nifurtimox
(3-methyl-4(5'-nitro-furfurylideneamino)-tetrahydro-4H-1,4-thiazine-1,1-dioxide)
is quite effective in treatment of acute Chagas' disease (Brener, 1979).
Nifurtimox (Bayer 2502;LAMPIT) is no longer commercially available but can be obtained in the
Antiprotozoal Effects Nifurtimox is trypanocidal against both the trypomastigote and
amastigote forms of T. cruzi. Concentrations of 1 Absorption, Fate, and Excretion Nifurtimox is well absorbed after oral administration, with peak
plasma levels observed after about 3.5 hours (Paulos et al., 1989).
Despite this, only low concentrations of the drug (10 to 20 Therapeutic Uses Nifurtimox is employed in the treatment of American trypanosomiasis
(Chagas' disease) caused by T. cruzi. Although the drug markedly
reduces the parasitemia, morbidity, and mortality from acute Chagas' disease,
it is ineffective in chronic stages of this infection. Moreover, only about
half of the patients who complete a course of therapy appear cured of the
parasitic infestation. Treatment with nifurtimox has no effect on
irreversible organ lesions. Whether or not the cardiomyopathy associated with
chronic disease actually reflects an autoimmune disease that is independent
of the presence of trypanosomes is debatable (seeUrbina, 1999). The
clinical response of the acute illness to drug therapy varies with geographic
region; parasite strains present in Nifurtimox is given orally. Adults with acute infection should receive 8 to 10 mg/kg daily in four divided doses for 90 to 120 days. Children 1 to 10 years of age with acute Chagas' disease should receive 15 to 20 mg/kg per day in four divided doses for 90 days; for individuals 11 to 16 years old, the daily dose is 12.5 to 15 mg/kg given according to the same schedule. Gastric upset and weight loss can occur during treatment. If the latter occurs, dosage should be reduced. The ingestion of alcohol should be avoided during treatment, because the incidence of side effects may increase. Toxicity and Side Effects Children tolerate nifurtimox better than do adults. Nonetheless, drug-related side effects are common. They range from hypersensitivity reactionssuch as dermatitis, fever, icterus, pulmonary infiltrates, and anaphylaxisto dose- and age-dependent complications primarily referable to the gastrointestinal tract and both the peripheral and central nervous systems (seeBrener, 1979). Nausea and vomiting are common, as are myalgia and weakness. Peripheral neuropathy and gastrointestinal symptoms are especially common after prolonged treatment; the latter complication may lead to weight loss and preclude further therapy. Headache, psychic disturbances, paresthesias, polyneuritis, and CNS excitability are less frequent. Leukopenia and decreased sperm counts also have been reported. The compound may suppress cell-mediated immune reactions, both in vitro and in vivo (Lelchuk et al., 1977a,b). Because of the seriousness of Chagas' disease and the lack of superior drugs, there are few absolute contraindications to the use of nifurtimox. |
Pentamidine
|
History The discovery of antiprotozoal activity in the diamidine family of drugs was a fortuitous consequence of the search for hypoglycemic compounds that might compromise parasite energy metabolism. Of the compounds tested, three were found to possess outstanding activity: stilbamidine, pentamidine, and promamidine. Pentamidine was the most useful clinically because of its relative stability, lower toxicity, and ease of administration. Although it is effective clinically against a number of pathogenic protozoa, including Leishmania species, pentamidine is now used primarily for the prophylaxis and treatment of pulmonary and systemic infections with P. carinii in patients who cannot tolerate trimethoprim-sulfamethoxazole. It continues to be used alone or combined with suramin for the treatment of early-stage West African trypanosomiasis (seePpin and Milord, 1994; Ppin and Khonde, 1996). Diminazene (BERENIL) is a related diamidine that is used as a cheap alternative to pentamidine for the treatment of early African trypanosomiasis in some endemic areas, despite the fact that it is approved for veterinary use only. A number of promising analogs of pentamidine have been tested in a rat model of P. carinii infection (seeVhringer and Arasth, 1993), but none has been developed for human use. Chemistry Pentamidine has the following chemical structure:
Pentamidine isethionate is the preparation used clinically. It is marketed for injection (PENTAM 300) or for use as an aerosol (NEBUPENT). One milligram of pentamidine base is equivalent to 1.74 mg of the pentamidine isethionate. Solutions should be used promptly after preparation. Antiprotozoal and Antifungal Effects The positively charged aromatic diamidines are toxic to a number of different protozoa yet show rather marked selectivity of action. For example, the drugs are curative against T.b. rhodesiense and T.b. congolense infections in experimental animals but are ineffective in curing mice infected with T. cruzi. They also are capable of curing Babesia canis infections in puppies and Leishmania donovani infections in hamsters. These findings provide the basis for diamidine treatment of African trypanosomiasis and leishmaniasis in human beings. The diamidines also are fungicidal. Activity in vitro against Blastomyces dermatitidis led to the successful therapeutic trial of these drugs in systemic blastomycoses. The use of amphotericin B, however, has reduced the value of the diamidines in the treatment of this disease. At near therapeutic levels, pentamidine kills nonreplicating forms of P. carinii in culture (Pifer et al., 1983), but other evidence suggests that pentamidine exerts a biostatic rather than biocidal effect (seeVhringer and Arasth, 1993). Mechanism of Action and Resistance The mechanism of action of the diamidines is unknown. These dicationic compounds may display multiple effects on a given parasite and act by disparate mechanisms in different parasites (seeSands et al., 1985; Wang, 1995; Barrett and Fairlamb, 1999). In T. brucei, for example, the diamidines are concentrated via an energy-dependent, high-affinity uptake system that operates more effectively in drug-sensitive than in drug-resistant strains (Damper and Patten, 1976). The diamidines utilize a transporter selective for adenine and adenosine, purines that must be imported to assure parasite survival (Barrett and Fairlamb, 1999). Melamine-based arsenicals use the same purine (P-2) transporter, which explains the cross-resistance to diamidines exhibited by certain arsenical-resistant strains of T. brucei (seeCarter et al., 1995; Barrett and Fairlamb, 1999; Maser et al., 1999; de Koning and Jarvis, 1999). Although failure to concentrate diamidines is the usual cause of pentamidine resistance, other mechanisms could be involved (Berger et al., 1995). After achieving millimolar concentrations within trypanosomes, the positively charged hydrophobic diamidines may exert their trypanocidal effects by reacting with a variety of negatively charged intracellular targets such as membrane phospholipids, enzymes, RNA, and DNA. Indeed, ribosomal aggregation, inhibition of DNA and protein synthesis, and inhibition of several enzymesalong with seeming loss of trypanosomal kinetoplasthave all been reported (seeBarrett and Fairlamb, 1999). Inhibition of S-adenosyl-L-methionine decarboxylase in vitro suggested that pentamidine might interfere with polyamine biosynthesis, but this seems unlikely to explain the drug's action in vitro (Bitoni et al., 1986; Berger et al., 1993). Inhibition in vitro of trypanosomal mitochondrial topoisomerase II and plasma Ca2+,Mg2+-ATPase also has been reported (seeBarrett and Fairlamb, 1999). The diamidines bind to DNA at sequences composed of at least four consecutive A-T base pairs (Bailly et al., 1994). Pentamidine promotes linearization of trypanosome kinetoplast DNA, consistent with its being a type II topoisomerase inhibitor (Shapiro and Englund, 1990). The drug also inhibits ATP-dependent topoisomerases in extracts of P. carinii (Dykstra and Tidwell, 1991). Absorption, Fate, and Excretion The pharmacokinetics and biodisposition of pentamidine isethionate have been studied most extensively in AIDS patients with P. carinii infections (seeVhringer and Arasth, 1993); information from patients with Gambian trypanosomiasis is more limited (seePpin and Milord, 1994; Bronner et al., 1995). Pentamidine isethionate is fairly well absorbed from parenteral sites of administration despite the formation of sterile abscesses that may occur after its use. Following a single intravenous dose, the drug disappears from plasma with an apparent half-life of several minutes to a few hours; this is followed by a slower distribution phase and a prolonged elimination phase lasting from weeks to months. Patients with African trypanosomiasis exhibit marked interindividual variations in pharmacokinetic parameters. Their mean system plasma clearance after a single dose is about 1120 ml/minute, but the volume of distribution is about 25,000 liters, a finding that accounts for the prolonged average elimination half-life of about 12 days (Bronner et al., 1995). The renal clearance of pentamidine averages only about 2% to 11% of its systemic clearance (Conte, 1991; Bronner et al., 1995), but whether the drug is metabolized or excreted in bile, for example, is unknown. In patients receiving multiple injections of the drug over a 13-day period for treatment of pneumocystosis, drug accumulation occurs such that no steady-state plasma concentration is attained (Conte, 1991). Extensive accumulation of pentamidine in tissues and its slow excretion during repeated administration may relate to both its therapeutic properties and its prophylactic efficacy in both pneumocystosis and African trypanosomiasis (seePpin and Milord, 1994). After multiple parenteral doses, the liver, kidney, adrenal, and spleen of patients with AIDS contain the highest concentrations of drug, whereas only traces are found in the brain (Donnelly et al., 1988). Lungs of such patients contain intermediate but therapeutic concentrations after five daily doses of 4 mg of base per kilogram. Higher pulmonary concentrations should be achieved by inhalation of pentamidine aerosols for prophylaxis or as adjunctive treatment for mild to moderate P. carinii pneumonia; delivery of drug by this route results in little systemic absorption and decreased toxicity compared with intravenous administration in both adults and children (Leoung et al., 1990; Hand et al., 1994). The actual dose delivered to the lungs depends on both the size of particles generated by the nebulizer and the patient's ventilatory patterns. Therapeutic Uses Pentamidine isethionate usually is given by intramuscular injection or by slow intravenous infusion over 60 minutes in single daily doses of 4 mg of base per kilogram. However, dosage regimens vary by disease and, in some instances, are not firmly established (see above). For treatment of early lymphatic African trypanosomiasis due to T.b. gambiense, pentamidine can be given intramuscularly on days 1, 3, 5, 7, 13, and 17, while suramin is administered intravenously (20 mg/kg up to a maximum of 1 g) on days 1 and 13. An alternative is to give seven intramuscular doses of pentamidine alone on alternate days (seePpin and Milord, 1994). Because of failure to penetrate the CNS, pentamidine is not used to treat T.b. rhodesiense, which affects the brain early in the course of infection. The drug also is largely ineffective in T.b. gambiense infections once the CNS is involved. Pentamidine has been used successfully in courses of 12 to 15 intramuscular doses of 2 to 4 mg/kg, either daily or every other day, to treat visceral leishmaniasis (kala azar caused by L. donovani). This compound provides an alternative to antimonials or lipid formulations of amphotericin B for patients who cannot tolerate the latter agents. Pentamidine isethionate given as 4 intramuscular doses of 3 mg/kg every other day has enjoyed some success in the treatment of cutaneous leishmaniasis (Oriental sore caused by L. tropica) but is not used routinely to treat this infection (seeBerman, 1997). Pentamidine is one of a number of drugs and drug combinations used extensively for the prophylaxis and treatment of infections with P. carinii, P. carinii pneumonia (PCP) being the most common opportunistic infection in individuals infected with HIV. In western countries, these drugs have markedly reduced mortality from PCP, changed the spectrum of AIDS-related illnesses, and increased life expectancy in this population. Fauci and Lane (1998) have reviewed the use of pentamidine and other drugs for the prophylaxis and treatment of P. carinii infections in HIV-infected adults. Prophylaxis against PCP is recommended for such persons with either a previous P. carinii infection, a CD4 lymphocyte count of 200 cells per microliter or less, an unexplained fever for 2 or more weeks, or a history of oropharyngeal candidiasis. Pentamidine, taken by aerosol inhalation, is no longer recommended for routine prophylaxis against PCP; instead, it is reserved for those few individuals unable to tolerate systemic therapy with more effective agents, e.g., trimethoprim-sulfamethoxazole (seeChapter 44: Antimicrobial Agents: Sulfonamides, Trimethoprim-Sulfamethoxazole, Quinolones, and Agents for Urinary Tract Infections). The usual monthly dose is 300 mg of a 5% to 10% nebulized aqueous solution of pentamidine isethionate delivered over 30 to 45 minutes via a RESPIRGARD II device. Disadvantages of aerosolized pentamidine are that it lacks efficacy against extrapulmonary infections with P. carinii and it induces an increased incidence of pneumothorax. For treatment of moderate to severe PCP or systemic pneumocystosis in nondebilitated, HIV-infected adults, pentamidine isethionate is the preferred alternative to trimethoprim-sulfamethoxazole when the latter cannot be tolerated. The optimal dosage regimen is 4 mg/kg of pentamidine given intravenously each day for 21 days, depending on the severity of infection and tolerance to the drug. In some instances, a lower 2- to 3-mg/kg daily dosage may be equally effective and produce substantially less toxicity (seeVhringer and Arasth, 1993). HIV-infected patients with PCP may worsen during the first 5 days of therapy, possibly from an inflammatory response to killed organisms. Treatment with corticosteroids may ameliorate this life-threatening response if instituted as soon as the diagnosis is made but no later than 72 hours thereafter. Clinical improvement usually occurs by the end of the first week of pentamidine therapy if the patient responds. A high proportion of cures can be expected even though side effects may force cessation of therapy. The prognosis is less favorable in debilitated patients with altered immunity or neoplastic disease, who may require more than one course of therapy. Consistent with a biostatic action of pentamidine against P. carinii, treatment failures, relapses, and drug toxicity and intolerance are especially prevalent in patients with AIDS. These individuals are more likely to respond favorably to trimethoprim-sulfamethoxazole if that medication is tolerated. The use of pentamidine has reduced mortality markedly in the epidemic form of P. carinii infection found in debilitated and premature infants. While the feasibility of administering this drug by aerosol to infants with HIV infection has been demonstrated, the efficacy of such therapy has yet to be proven (Hand et al., 1994). Toxicity and Side Effects At therapeutic doses (4 mg/kg per day), pentamidine causes toxicity in about 50% of patients treated, whether or not they have AIDS. The major complications of pentamidine therapy have been well reviewed (seeSands et al., 1985; Vhringer and Arasth, 1993). Intravenous injection of pentamidine (and other diamidines) can be followed by alarming and sometimes dangerous reactions. These include breathlessness, tachycardia, dizziness or fainting, headache, and vomiting. These reactions probably relate to the sharp fall in blood pressure that follows too rapid intravenous administration of the drug, and they may be due in part to the release of histamine. If solutions of pentamidine cannot be given slowly by the intravenous route, the drug is well tolerated after intramuscular injection. However, the latter route is associated with formation of sterile abscesses at the injection site. Pentamidine does not appear to cause late neuropathies. Pancreatitis and hypoglycemia and, paradoxically, hyperglycemia and insulin-dependent diabetes have been documented following its administration; the hypoglycemia may be life-threatening or even fatal if not recognized (seeSands et al., 1985). Other adverse effects include skin rashes, thrombophlebitis, thrombocytopenia, anemia, neutropenia, elevation of liver enzymes, and nephrotoxicity (seeVhringer and Arasth, 1993). Impaired renal function has been seen in 24% of patients receiving the drug, but this is usually reversible. |
Quinacrine
|
Quinacrine is an acridine derivative widely
used during World War II as an antimalarial agent. Although it has been
replaced by newer and safer antimalarial drugs (seeChapter 40: Drugs
Used in the Chemotherapy of Protozoal Infections: Malaria), quinacrine
hydrochloride is very effective against G. lamblia, producing cure
rates of at least 90%. However, quinacrine is no longer available in the |
Sodium Stibogluconate
|
History Antimonial compounds long have been used for therapy of leishmaniasis and other protozoal infections. The first trivalent antimonial compound used to treat cutaneous leishmaniasis and kala azar was antimony potassium tartrate (tartar emetic), which was both toxic and difficult to administer. Tartar emetic and other trivalent arsenicals eventually were replaced by pentavalent antimonial derivatives of phenylstibonic acid. These drugs were as effective as but far less toxic than tartar emetic, thus permitting the use of larger doses and shorter periods of treatment. Later syntheses reverted to the 'tartar emetic' type of compound in which trivalent antimony was replaced by pentavalent antimony. An early member of this type of compound was sodium stibogluconate (sodium antimony gluconate;PENTOSTAM). This drug is widely used today and, together with meglumine antimonate (GLUCANTIME), a pentavalent antimonial compound preferred in French-speaking countries, is the mainstay of the treatment of leishmaniasis by antimony. The history of leishmanicides has been reviewed by Steck (1974) and Berman (1988). Chemistry Sodium stibogluconate has the following chemical structure:
Clinical formulations of sodium stibogluconate actually consist of
multiple uncharacterized molecular forms, some of which have higher molecular
masses than the compound shown (seeBerman, 1988). Typical preparations
contain 30% to 34% pentavalent antimony by weight as well as m-chlorocresol
added as a preservative. In the Antiprotozoal Effects and Drug Resistance The mechanism of the antileishmanial action of sodium stibogluconate remains to be clarified. Exposure to this compound compromises the bioenergetics of Leishmania amastigotes (seeBerman, 1988). Both glycolysis and fatty acid metabolism, processes primarily localized in unusual organelles termed glycosomes, are suppressed. Sodium stibogluconate also diminishes net generation of ATP and GTP. Whether this is a primary or a secondary drug effect is not known. Other mechanisms such as carbohydrate-directed targeting of the drug to macrophages and nonspecific binding of antimony to the sulfhydryl groups of amastigote proteins may be involved (Roberts et al., 1995). Recent studies support the old hypothesis that relatively nontoxic pentavalent antimonials act as prodrugs. These compounds are reduced to the more toxic Sb3+ species that kill amastigotes within the phagolysosomes of macrophages (Roberts et al., 1995; Sereno and Lemesre, 1997; Sereno et al., 1998). Incidentally, but perhaps importantly, the m-chlorocresol preservative used in formulations of sodium stibogluconate has intrinsic activity that may contribute to antileishmanial effects of the drug preparation in vivo (Roberts and Rainey, 1993; Sereno et al., 1998). Naturally resistant Leishmania or parasites made resistant to antimonial drugs in culture show several biochemical differences from drug-sensitive strains. Resistant strains have displayed elevated levels of intracellular thiols, especially trypanothione, and amplification of the ATP-binding cassette (ABC) transporter gene, pgp A. However, while these changes may contribute to antimonial drug resistance, they are variable and insufficient to confer the high degree of resistance noted for some strains (seeLegare et al., 1997; Haimeur et al., 2000). Indeed, susceptibility of L. donovani to pentavalent antimonials appears to be parasite-intrinsic, stage-specific (amastigotes are more susceptible than promastigotes), and macrophage-independent (Ephros et al., 1999). Absorption, Fate, and Excretion The pentavalent antimonials attain much higher concentrations in plasma than do the trivalent compounds. Consequently, most of a single dose of sodium stibogluconate is excreted in the urine within 24 hours. Its pharmacokinetic behavior is similar whether the drug is given intravenously or intramuscularly (Pamplin et al., 1981; Chulay et al., 1988). The agent is rapidly absorbed, distributed in an apparent volume of about 0.22 liter/kg, and eliminated in two phases. The first has a short half-life of about 2 hours, and the second is much slower (t1/2= 33 to 76 hours). The prolonged terminal elimination phase may reflect conversion of the pentavalent antimonial (Sb5+) to the more toxic trivalent (Sb3+) form that is concentrated and slowly released from tissues. Indeed, about 20% of the plasma antimony is present in the trivalent form after pentavalent antimonial administration. Sequestration of antimony in macrophages also may contribute to the prolonged antileishmanial effect after plasma antimony levels have dropped below the minimal inhibitory concentration (MIC) observed in vitro. Therapeutic Uses The changing use of sodium stibogluconate, meglumine antimonate, and
other agents for the chemotherapy of leishmaniasis has been extensively
reviewed (seeBerman, 1988 and 1997). Sodium stibogluconate is given
either intravenously or intramuscularly, the dosage regimen depending on the
local responsiveness of a particular form of leishmaniasis to this compound.
Prolonged dosage schedules with maximally tolerated doses are now needed for
successful therapy of visceral, mucosal, and some forms of cutaneous
leishmaniasis, in part to overcome increasing clinical resistance to
antimonial drugs. Even high-dose regimens may no longer produce satisfactory
results. For example, pentavalent antimonials are no longer superior to amphotericin
B for treatment of either visceral leishmaniasis (kala azar) in The current recommended daily dose of sodium stibogluconate has been
increased from 10 to 20 mg of pentavalent antimony per kilogram body weight,
a dose that ordinarily causes minimal added risk to patients. Given
intramuscularly for 10 days, this regimen has cured over 90% of individuals
with cutaneous leishmaniasis. For visceral leishmaniasis, except in The incidence of treatment failures with sodium stibogluconate in visceral, mucocutaneous, and some forms of cutaneous leishmaniasis is increasing dramatically in endemic areas (Ouellette and Papadopoulou, 1993). Although many treatment failures may be attributable to reinfection or to pharmacokinetic or immunological variability in patients, resistance to sodium stibogluconate is well documented in both laboratory-derived strains and clinical isolates (Berman et al., 1989; Grogl et al., 1992). Indeed, assays of intracellular amastigote resistance to sodium stibogluconate in vitro may prove helpful in predicting the clinical response of visceral L. donovani infections to this drug (Lira et al., 1999). However, sodium stibogluconate still remains the drug of choice for leishmaniasis. Its main disadvantages are the long courses of therapy required, the necessity for parenteral administration, and its relatively high cost. For cases of East African and Indian kala azar and mucosal leishmaniasis that are unresponsive to pentavalent antimonials, lipid formulations of amphotericin B are preferred. Toxicity and Side Effects The toxicity of the pentavalent antimonials is best evaluated in patients without systemic disease, i.e., visceral leishmaniasis. In general, high-dose regimens of sodium stibogluconate are fairly well tolerated; toxic reactions are usually reversible, and most subside despite continued therapy. Effects most commonly noted include pain at the injection site after intramuscular administration; chemical pancreatitis in nearly all patients; elevation of serum hepatic transaminase levels; bone-marrow suppression manifested by decreased red-cell, white-cell, and platelet counts in the blood; muscle and joint pain; weakness and malaise; headache; nausea and abdominal pain; and skin rashes. Changes in the electrocardiogram that include T-wave flattening and inversion and prolongation of the QT interval found in patients with systemic disease are uncommon in other forms of leishmaniasis (Navin et al., 1992; Berman, 1997; Sundar et al., 1998). Reversible polyneuropathy has been reported. Hemolytic anemia and renal damage are rare manifestations of antimonial toxicity, as are shock and sudden death. |
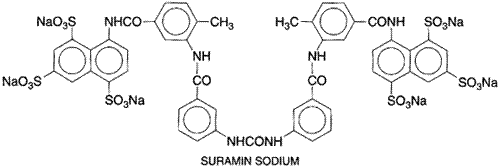
Suramin
|
History Based on the trypanocidal activity of the dyestuffs trypan red,
trypan blue, and afridol violet, several years of research in Chemistry and Preparation Suramin sodium BAYER 205, formerly GERMANIN, others) has the chemical structure shown here.
The drug is soluble in water, but solutions deteriorate quickly in
air; only freshly prepared solutions should be used. In the Antiparasitic Effects Suramin is a relatively slowly acting trypanoside (>6 hours in vitro) with high clinical activity against both T.b. gambiense and T.b. rhodesiense and an unknown mechanism of action (seeVoogd et al., 1993). Its delayed onset of action probably stems from slow endocytic uptake of the drug-protein complex by trypanosomes (Fairlamb and Bowman, 1977). Structural modifications of this polyanion usually result in substantial loss of trypanocidal activity. Suramin reacts reversibly with a variety of biomolecules in vitro, inhibiting many trypanosomal and mammalian enzymes unrelated to its antiparasitic effects (seePpin and Milord, 1994; Wang, 1995 and 1997). Compartmentation protects many vital molecules, such as the glycolytic enzymes inside trypanosomal glycosomes, from suramin's action, because this compound does not cross membrane barriers by passive diffusion (Wang, 1995). However, the chemical structure of suramin may confer transport specificity, because removal of its two methyl groups results in the loss of trypanocidal activity in vivo but not in vitro (Morty et al., 1998). Suramin-treated trypanosomes exhibit damage to intracellular membrane structures other than lysosomes, but whether or not this relates to the drug's primary action is unknown. Inhibition of a trypanosomal cytosolic serine oligopeptidase may account for at least part of suramin's activity (Morty et al., 1998). Suramin is the only microfilaricide used clinically, albeit rarely now, for treatment of human onchocerciasis (seeChapter 42: Drugs Used in the Chemotherapy of Helminthiasis). This compound displays delayed but prolonged filaricidal activity against both adult male and female worms and lesser but significant activity against microfilariae. Its mechanism of action against Onchocerca volvulus is unknown (seeVoogd et al., 1993). Absorption, Fate, and Excretion Because it is not absorbed after oral intake, suramin is given intravenously to avoid local inflammation and necrosis associated with subcutaneous or intramuscular injections. After its administration, the drug displays complex pharmacokinetics with marked interindividual variability. The concentration in plasma falls fairly rapidly for a few hours, more slowly for a few days, and finally very slowly, with a terminal elimination half-life of about 90 days. The persistence of suramin in the circulation is due to extremely tight binding to plasma proteins; over 99.7% of the drug is bound after a typical 1-g dose (seePpin and Milord, 1994). The drug is not appreciably metabolized, and its renal clearance of about 5 ml/hour accounts for elimination of about 80% of the compound from the body. Although the apparent volume of distribution of suramin in an adult is about 20 liters, this large polar anion does not enter cells readily, and tissue concentrations are uniformly lower than those in plasma. In experimental animals, however, the kidneys contain considerably more suramin than do other organs. Such retention may account for the fairly frequent occurrence of albuminuria following injection of the drug in human beings. Very little suramin penetrates the cerebrospinal fluid, consistent with its lack of efficacy once the CNS has been invaded by trypanosomes. The dose-dependent, prolonged persistence of suramin in the circulation explains why the drug has been used for prophylaxis of African trypanosomiasis. Therapeutic Uses Suramin is used to treat African trypanosomiasis but is of no value in South American trypanosomiasis caused by T. cruzi. Because only small amounts of the drug enter the brain, suramin is used primarily to treat early stages (before CNS involvement) of both East and West African trypanosomiasis (seePpin and Milord, 1994). For therapy of early West African infections, this drug is more effective when given by intravenous regimens that also include intramuscular injections of pentamidine. In contrast, suramin alone appears superior for therapy of early East African disease. Suramin will clear the hemolymphatic system of trypanosomes even in late-stage disease, so it is often administered before initiating melarsoprol to reduce the risk of reactive encephalopathy associated with the administration of that arsenical (see above). In animal models, suramin has been found to display synergism with other trypanocides, including eflornithine. However, suramin-eflornithine therapy has been disappointing against late stage human T.b. rhodesiense infection (Clerinx et al., 1998). Suramin is given by slow intravenous injection as a 10% aqueous solution. Treatment of active African trypanosomiasis should not be started until 24 hours after diagnostic lumbar puncture, and caution is required if the patient has onchocerciasis because of the potential for eliciting a Mazzotti reaction. The normal single dose for adults with T.b. rhodesiense infection is 1 g. It is advisable to employ a test dose of 200 mg initially to detect sensitivity, after which the normal dose is given on days 1, 3, 7, 14, and 21. The pediatric dose is 20 mg/kg, given according to the same schedule. Patients in poor condition should be treated with lower doses during the first week. When suramin and pentamidine are used to treat early stage T.b. gambiense infection, Ppin and Milord (1994) recommend suramin (20 mg/kg, up to a maximum of 1 g) given intravenously on days 1 and 13, and pentamidine isethionate (4 mg/kg) given intramuscularly on days 1, 3, 5, 13, 15, and 17. However, suramin and pentamidine may lose their advantage over pentamidine alone for treatment of Gambian trypanosomiasis if there is even minimal evidence of CNS involvement (Ppin and Khonde, 1996). Patients who relapse after suramin therapy should be treated with melarsoprol (seePpin and Milord, 1994). Suramin is effective for the prophylaxis of African trypanosomiasis. Chemoprophylaxis is not recommended for travelers on occasional brief visits to endemic areas, because the risk of serious drug toxicity outweighs the risk of acquiring the disease (see below). For chemoprophylaxis, the single dose of 1 g is repeated weekly for 5 or 6 weeks. Toxicity and Side Effects Suramin can cause a variety of untoward reactions that vary in intensity and frequency and tend to be more severe in debilitated patients. Fortunately, the most serious immediate reaction consisting of nausea, vomiting, shock, and loss of consciousness is very rare (about 1 in 2000 patients). Malaise, nausea, and fatigue are common immediate reactions. Parasite destruction may cause febrile episodes and skin hypersensitivity rashes that are reduced by pretreatment with glucocorticoids; concomitant onchocerciasis optimally should be treated first with ivermectin to minimize these reactions (seeChapter 42: Drugs Used in the Chemotherapy of Helminthiasis). The most common problem encountered after several doses of suramin is renal toxicity, manifested by albuminuria; delayed neurological complications, including headache, metallic taste, paresthesias, and peripheral neuropathy also occur. These complications usually disappear spontaneously despite continued therapy. At higher doses over long periods of treatment used for cancer chemotherapy, suramin-induced coagulopathy is the most common toxicity observed, whereas development of a severe polyradiculoneuropathy is the most serious complication (seeVoogd et al., 1993; Bowden et al., 1996). Other less-prevalent reactions include vomiting, diarrhea, stomatitis, chills, abdominal pain, and edema. Laboratory abnormalities noted in 12% to 26% of patients with AIDS include leukopenia and occasional agranulocytosis, thrombocytopenia, proteinuria, and elevations of plasma creatinine, transaminases, and bilirubin, which are reversible. Unexpected findings in patients with AIDS include adrenal insufficiency and vortex keratopathy. Precautions and Contraindications Patients receiving suramin should be followed closely. Therapy should not be continued in patients who show intolerance to initial doses, and the drug should be employed with great caution in individuals with renal insufficiency. Moderate albuminuria is common during control of the acute phase, but persisting, heavy albuminuria calls for caution as well as modification of the treatment schedule. If casts appear, treatment with suramin should be discontinued. The occurrence of palmar-plantar hyperesthesia may presage peripheral neuritis. |
Antiprotozoal Antibiotics
|
Numerous antibiotics have been tested for efficacy against many protozoal infections. A few of these have been shown to be of benefit, but not usually as primary drugs. Whereas tetracycline is recommended as first-line therapy for balantidiasis and intestinal infections with Dientamoeba fragilis, clindamycin is prescribed along with quinine for therapy of babesiosis. Paromomycin is discussed below because it has clinical efficacy against a wide spectrum of protozoal infections. Unlike some tetracyclines and erythromycin, which also have been used to treat intestinal amebiasis, paromomycin is the only antibiotic that is directly amebicidal. Other antibiotics act by interfering with the enteric flora essential for proliferation of pathogenic amoebae. Paromomycin This aminoglycoside antibiotic, isolated from cultures of Streptomyces
rimosus, is structurally related to neomycin and shares most of the
antibacterial properties of other antibiotics in this class (seeChapter
46: Antimicrobial Agents: The Aminoglycosides). Indeed, paromomycin is
licensed in
The recommended dosage of paromomycin sulfate (HUMATIN) for intestinal amebiasis is 25 to 35 mg/kg given orally in three divided doses at mealtimes for 7 days. Higher dosages, up to 66 mg/kg, have been used by some practitioners. After oral administration, little of the drug is absorbed into the systemic circulation. Side effects are limited mainly to gastrointestinal upset and diarrhea. Marked renal damage occurs in animals treated parenterally with this drug. Human beings who are given injectable formulations of paromomycin may experience damage to both the kidney and the eighth cranial nerve if the recommended dosage is exceeded or if they also are exposed to other potentially toxic agents (seeBerman, 1997). Experience has shown paromomycin to be effective but by no means infallible in the treatment of intestinal amebiasis; it is ineffective against extraintestinal forms of the disease (seeWoolfe, 1965). |
Prospectus
|
Two major challenges must be faced to improve the chemoprophylaxis and therapy of human protozoal infections. The first is to find effective remedies for infections, such as Chagas' disease and East African trypanosomiasis, that respond poorly if at all to known drugs. The second is to deal successfully with increased inherent and acquired resistance of pathogenic protozoa to effective antiprotozoal agents. Examples here include increasing resistance of T. brucei to melarsoprol, Leishmania to pentavalent antimonials, and T. vaginalis to metronidazole. Progress in finding new antiprotozoal drugs and combating drug resistance has been hampered by lack of economic incentives and commitment to emphasize research and drug development in this field. Several strategies may help address these problems despite economic limitations. To avoid some of the high costs associated with drug development and regulatory approval, drugs already approved for other indications could be tested for efficacy against human protozoal diseases in limited clinical trials. Ideally, this decision would be based on supportive evidence from in vitro experiments and animal models. Several examples illustrate the utility of this approach. Atovaquone, approved for treatment of infections with P. carinii, is now used clinically against multidrug-resistant falciparum malaria and infections with T. gondii. Eflornithine, originally evaluated as an antineoplastic agent, is clinically effective against West African trypanosomiasis. Miltefosine, an alkylphospholipid originally tested for safety as an anticancer drug, shows promise as the first oral medication for treatment of visceral leishmaniasis (seeHerwaldt, 1999a). Drugs developed to combat protozoal infections in animals provide a rich source of possible candidates for human use; such compounds often are cheap and readily available. Diminazine (BERENIL) is a case in point. This aromatic diamidine used to treat bovine trypanosomiasis has shown some promise for the therapy of East African trypanosomiasis. However, there is little economic incentive to develop it for human use. Combination chemotherapy with known effective antiprotozoal agents is another approach that can prove fruitful. Ideally, a combination would be selected on the basis of known complementary antiprotozoal activities, just as was done with the antifolates. Alternatively, drugs acting on different but vital protozoal processes also may enhance each other's effect. This approach, along with the use of drugs with compatible pharmacokinetic properties, has proven to be successful, not only to improve the clinical response but also to circumvent and delay the emergence of drug resistance. Several multidrug regimens for the treatment of multidrug-resistant falciparum malaria amply illustrate this point (seeChapter 40: Drugs Used in the Chemotherapy of Protozoal Infections: Malaria). The effectiveness of antiprotozoal chemotherapy is critically
dependent on the status of host defense mechanisms. The worldwide AIDS
epidemic is notoriously associated with exacerbations of many coexisting
infections, including those with pathogenic protozoa. For example, visceral
leishmaniasis as a component of AIDS has increased dramatically in southern Targeted drug delivery represents yet another strategy to combat protozoal infections. This alternative is largely experimental and, as yet, too costly to be of much practical use. For instance, associating pentavalent antimony with mannan instead of sodium gluconate increases its potency against Leishmania amastigotes in culture, possibly by directing the antimony complexes to macrophages (Roberts et al., 1995). Moreover, newer lipid formulations of amphotericin B have increased the therapeutic index of this antifungal agent, and it is now considered first-line therapy for Indian visceral leishmaniasis. The best long-term defense against protozoan infections, however, is continued basic and clinical research directed at understanding parasite biology as it applies to the rational design and development of improved drugs and vaccines. Although the cost of this effort is high, the cost of failure is much higher. |
|
Politica de confidentialitate | Termeni si conditii de utilizare |

Vizualizari: 2665
Importanta: ![]()
Termeni si conditii de utilizare | Contact
© SCRIGROUP 2026 . All rights reserved